Синдром бругада код мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Бругада.
Синдром Бругада
Описание
Синдром Бругада. Генетически обусловленное кардиологическое состояние, характеризующееся различными нарушениями работы сердца, которые приводят к резкому повышению риска развития внезапной сердечной смерти. Симптомами этого состояния являются приступы пароксизмальной тахикардии, обмороки, фибрилляции предсердий и жизнеугрожающие фибрилляции желудочков, чаще всего возникающие во время сна. Диагностика синдрома Бругада производится на основании характерного симптомокомплекса, электрокардиографических данных и изучения наследственного анамнеза, некоторые формы патологии определяются молекулярно-генетическими методами. Специфического лечения заболевания не существует, применяют антиаритмическую терапию, используют разнообразные кардиостимуляторы.
Дополнительные факты
Синдром Бругада – группа генетических нарушений, приводящих к изменению ионной проницаемости мембран кардиомиоцитов, вследствие чего возникают патологии ритма и проводимости, создающие повышенный риск внезапной сердечной смерти. Впервые такое состояние было описано в 1992 году двумя братьями – бельгийскими кардиологами испанского происхождения Хосе и Педро Бругада, обратившими внимание на взаимосвязь определенных электрокардиологических проявлений и нарушений сердечного ритма. В настоящее время установлено, что синдром Бругада является наследственным состоянием с предположительно аутосомно-доминантным механизмом передачи, удалось выявить несколько генов, мутации которых способны вызывать это заболевание. По некоторым данным, практически половина всех случаев внезапной сердечной смерти в мире обусловлены именно этой патологией. Распространенность синдрома Бругада различается в разных регионах планеты – в странах Америки и Европы она составляет примерно 1:10 000, тогда как в африканских и азиатских государствах это заболевание встречается чаще – 5-8 случаев на 10 000 населения. Синдром Бругада примерно в 8 раз чаще поражает мужчин, чем женщин, проявления патологии возникают в разном возрасте, но чаще всего выраженная симптоматика наблюдается в 30-45 лет.
Синдром Бругада
Причины
Причина развития нарушений при синдроме Бругада заключается в патологической работе ионных каналов кардиомиоцитов, в основном натриевых и кальциевых. Их дефект, в свою очередь, обусловлен мутациями генов, кодирующих белки ионных каналов. Методами современной генетики удалось достоверно идентифицировать 6 основных генов, поражение которых приводит к развитию синдрома Бругада, в отношении еще нескольких существует подозрение, но отсутствует необходимая доказательная база. На этой основе построена классификация данного состояния, включающая в себя 6 форм заболевания (BrS):
• BrS. 1 — наиболее распространенный и хорошо изученный вариант синдрома Бругада. Обусловлен мутацией гена SCN5A, расположенного на 3 хромосоме. Продуктом экспрессии данного гена является альфа-субъединица натриевого канала 5 типа, широко представленного в миокарде. Помимо синдрома Бругада мутации данного гена становятся причиной большого количества наследственных кардиологических патологий – семейной фибрилляции предсердий, синдрома слабости синусового узла 1 типа и ряда других.
• BrS. 2 — данная разновидность синдрома Бругада вызывается дефектами гена GPD1L, который локализован на 3 хромосоме. Он кодирует один из компонентов глицерол-3-фосфат дегидрогеназы, принимающей активное участие в работе натриевых каналов кардиомиоцитов.
• BrS. 3 — этот тип синдрома Бругада обусловлен дефектом гена CACNA1C, расположенного на 12 хромосоме. Продуктом его экспрессии является альфа-субъединица кальциевого канала L-типа, также присутствующего в кардиомиоцитах.
• BrS. 4 — как и в предыдущем случае, причиной развития синдрома Бругада 4 типа является поражение потенциал — зависимых кальциевых каналов L — типа. Оно обусловлено мутацией гена CACNB2, расположенного на 10 хромосоме и кодирующего бета-2-субъединицу вышеуказанных ионных каналов.
• BrS. 5 — распространенная разновидность синдрома Бругада, обусловленная мутацией гена SCN4B, локализованного на 11 хромосоме. Он кодирует белок одного из малых натриевых каналов кардиомиоцитов.
• BrS. 6 — вызывается дефектом гена SCN1B, расположенного на 19 хромосоме. Во многом этот вариант синдрома Бругада схож с первым типом заболевания, поскольку в этом случае тоже поражаются натриевые каналы 5 типа. Ген SCN1B кодирует бета-1-субъединицу этого ионного канала.
Кроме того, в развитии синдрома Бругада подозреваются мутации генов KCNE3, SCN10A, HEY2 и некоторых других. Однако на сегодняшний день достоверно доказать их роль в возникновении данного заболевания не удается, поэтому пока количество генетических вариантов синдрома Бругада ограничено шестью. Наследование всех форм данной патологии неясно, лишь у 25% больных определяются признаки аутосомно-доминантной передачи. Предположительно имеет место доминантный тип наследования с неполной пенетрантностью либо влияние спонтанных мутаций. Также непонятны причины того, почему синдром Бругада чаше поражает мужчин, нежели женщин – возможно, выраженность проявлений заболевания находится в зависимости от гормонального фона больного.
Классификация
Патогенез нарушений при любой форме синдрома Бругада примерно одинаков – из-за изменения проницаемости мембраны кардиомиоцитов для ионов натрия происходит нарушение трансмембранного потенциала и взаимосвязанных с ним характеристик возбудимых тканей: возбудимости, сократимости, передачи возбуждения окружающим клеткам. В результате возникают блокады проводящих путей сердца (пучков Гиса), тахиаритмии, усиливающиеся при повышении вагусных воздействий (во время сна). Степень выраженности симптомов при синдроме Бругада зависит от доли пораженных натриевых каналов. Усиливать проявления болезни могут некоторые лекарственные вещества, способны ингибировать ионные каналы сердца.
Симптомы
Возраст появления первых признаков синдрома Бругада сильно отличается у разных больных – были зарегистрированы случаи этой патологии как у детей 3-4 лет, так и у лиц старческого возраста. Одним из первых проявлений патологии становятся изменения на электрокардиограмме при полном отсутствии других клинических симптомов, поэтому данное заболевание нередко выявляется случайно. В большинстве случаев выраженная клиника синдрома Бругада возникает в возрасте 30-45 лет, этому предшествует бессимптомный период продолжительностью 10-12 лет, в течение которого единственным признаком патологии являются изменения на ЭКГ.
Диагностика
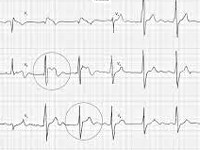
Для определения синдрома Бругада используют электрокардиографические методики, изучение наследственного анамнеза пациента, молекулярно-генетический анализ. Заподозрить наличие этого заболевания можно при наличии синкопальных явлений (головокружений, обмороков) неясного происхождения, жалоб на внезапные приступы тахиаритмий. Изменения на электрокардиограмме при синдроме Бругада могут определяться на фоне полного отсутствия клинических симптомов заболевания. При этом кардиологи выделяют три основных типа изменений на ЭКГ, незначительно отличающихся между собой. Типичная картина электрокардиограммы при синдроме Бругада сводится к элевации (подъему) сегмента ST над изоэлектрической линией и отрицательному зубцу Т на правых грудных отведениях (V1-V3). Также могут определяться признаки блокады правой ножки пучка Гиса, при холтеровском мониторировании выявляются приступы пароксизмальной тахикардии или фибрилляции предсердий.
Как правило, наследственный анамнез больных синдромом Бругада отягощен – среди родственников или предков имеются случаи летальных исходов от сердечной недостаточности, смерти во сне или внезапной сердечной смерти. Этот факт, а также наличие вышеперечисленных симптомов и изменений на ЭКГ дает основания для проведения молекулярно-генетической диагностики. В настоящее время врачи-генетики в подавляющем большинстве клиник и лабораторий производят определение синдрома Бругада, вызванного только мутациями генов SCN5A и SCN4B (1 и 5 типы патологии), в отношении остальных форм методы генетической диагностики пока не разработаны.
Дифференциальная диагностика
Дифференцировать это состояние следует с реакцией организма на прием некоторых лекарственных средств, хроническим миокардитом и другими кардиологическими патологиями.
Лечение
Специфических методов лечения синдрома Бругада на сегодняшний момент не существует, поэтому ограничиваются только борьбой с проявлениями этого заболевания, а также профилактикой жизнеугрожающих приступов тахиаритмии и фибрилляций. Наиболее широко при этом состоянии применяется амиодарон, несколько реже используются дизопирамид и хинидин. Однако медикаментозная терапия при синдроме Бругада в ряде случаев является малоэффективной, единственным надежным средством профилактики аритмии и внезапной сердечной смерти в этом случае становится имплантация кардиовертера-дефибриллятора. Только этот прибор способен оценивать работу миокарда больного и при патологических и жизнеугрожающих изменениях сердечного ритма приводить ее в норму посредством электрического разряда.
Профилактика
Многие традиционные антиаритмические препараты при синдроме Бругада противопоказаны, так как они угнетают деятельность натриевых каналов кардиомиоцитов и усиливают проявления патологии. К средствам, запрещенным при этом заболевании, относят аймалин, пропафенон, прокаинамид. Поэтому больным синдромом Бругада следует обязательно сообщать специалистам об имеющемся диагнозе, чтобы избежать назначения неверного антиаритмического средства. При наличии подобного заболевания у родственников или случаях внезапной сердечной смерти в роду следует регулярно производить ЭКГ-исследование для как можно более ранней диагностики этого состояния.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Синдром Бругада — наследственное заболевание[1], обусловленное мутацией гена SCN5A, расположенного в плече p 3-й хромосомы, кодирующего биосинтез белковых субъединиц натриевого канала кардиомиоцитов.
Впервые это понятие, позже ставшее эпонимом, предложили испано-бельгийские кардиологи — братья Педро и Хосеп Бругада[2].
Мутации генов[править | править код]
На сегодняшний день известны, по крайней мере, 5 генов, ответственных за развитие этого состояния. В зависимости от мутации гена выделяют следующие варианты:
Патогенез[править | править код]
Заболевание имеет аутосомно-доминантный тип наследования в 25 % семей. Клинические проявления синдрома Бругада развиваются обычно в молодом возрасте (до 35-40 лет), реже — наблюдаются даже в пожилом и старческом возрасте. При исследовании статистических данных, накопленных в странах Юго-Восточной Азии и Дальнего Востока, было отмечено, что в данном регионе значительно распространены случаи внезапной ночной смерти в молодом возрасте (в год от 4 до 10 случаев на 10 000 жителей, в том числе в Лаосе — 1 случай на 10 000 жителей; в Таиланде — 26-38 на 100 000).
[7]
Описаны также случаи приобретенного синдрома Бругада[2].
Синдром Бругада характеризуется наличием преходящей полной или неполной блокады правой ножки пучка Гиса, косонисходящим подъёмом сегмента S-T в правых грудных отведениях (V1-V3), рецидивирующей пароксизмальной полиморфной желудочковой тахикардией и высоким риском внезапной сердечной смерти.
Симптомокомплекс[править | править код]
Полная форма синдрома Бругада включает следующие клинико-электрокардиографические проявления:
- Типичная электрокардиографическая картина (косонисходящее повышение сегмента S-T над изолинией на 1 мм и больше в отведениях V1—V3, на некоторых ЭКГ напоминает морду бультерьера, поэтому данное изменение иногда называют «типом бультерьера»[2], спонтанное или индуцированное введением антиаритмических препаратов I класса (блокаторов натриевых каналов, например, аймалина (гилуритмала) в дозе 1 мг/кг или новокаинамида в дозе 10 мг/кг, флекаинида 2 мг/кг); полная или неполная блокада правой ножки пучка Гиса); возможно укорочение интервала Q-T и удлинение P-Q (P-R);
- Пароксизмы полиморфной желудочковой тахикардии, часто рецидивирующие;
- синкопальные (обморочные) состояния;
- высокий риск внезапной сердечной смерти вследствие полиморфной желудочковой тахикардии или фибрилляции желудочков.
Примечания[править | править код]
- ↑ Окороков А. Н., Диагностика болезней внутренних органов: Т. 10. Диагностика болезней сердца и сосудов.: — М.: Мед. лит., 2005. — 384 с.: ил. ISBN 5-89677-091-X; — ст. 239—241.
- ↑ 1 2 3 Л. А. Бокерия, О. Л. Бокерия, Л. Н. Киртбая. СИНДРОМ БРУГАДА: КЛЕТОЧНЫЕ МЕХАНИЗМЫ И ПОДХОДЫ К ЛЕЧЕНИЮ. Анналы аритмологии, № 3, 2010.
- ↑ 1 2 Antzelevitch C; Pollevick GD; Cordeiro JM; Casis, O.; Sanguinetti, M. C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A. Loss-of-Function Mutations in the Cardiac Calcium Channel Underlie a New Clinical Entity Characterized by ST-Segment Elevation, Short QT Intervals, and Sudden Cardiac Death (англ.) // Circulation (англ.)русск. : journal. — Lippincott Williams & Wilkins (англ.)русск., 2007. — Vol. 115, no. 4. — P. 442—229. — doi:10.1161/CIRCULATIONAHA.106.668392. — PMID 17224476.
- ↑ Delpon E; Cordeiro JM; Núñez L; Thomsen, P. E. B.; Guerchicoff, A.; Pollevick, G. D.; Wu, Y.; Kanters, J. K.; Larsen, C. T. Functional Effects of KCNE3 Mutation and its Role in the Development of Brugada Syndrome (англ.) // Circulation Arrhythmia and Electrophysiology : journal. — 2008. — Vol. 1, no. 3. — P. 209—218. — doi:10.1161/CIRCEP.107.748103. — PMID 19122847.
- ↑ Watanabe H; Koopmann TT; Le Scouarnec S; Yang, Tao; Ingram, Christiana R.; Schott, Jean-Jacques; Demolombe, Sophie; Probst, Vincent; Anselme, Frédéric. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans (англ.) // Journal of Clinical Investigation (англ.)русск. : journal. — 2008. — June (vol. 118, no. 6). — P. 2260—2268. — doi:10.1172/JCI33891. — PMID 18464934.
- ↑ 1 2 Bezzina, Connie R; Barc, Julien; Mizusawa, Yuka; Remme, Carol Ann; Gourraud, Jean-Baptiste; Simonet, Floriane; Verkerk, Arie O; Schwartz, Peter J; Crotti, Lia; Dagradi, Federica; Guicheney, Pascale; Fressart, Véronique; Leenhardt, Antoine; Antzelevitch, Charles; Bartkowiak, Susan; Schulze-Bahr, Eric; Zumhagen, Sven; Behr, Elijah R; Bastiaenen, Rachel; Tfelt-Hansen, Jacob; Olesen, Morten Salling; Kääb, Stefan; Beckmann, Britt M; Weeke, Peter; Watanabe, Hiroshi; Endo, Naoto; Minamino, Tohru; Horie, Minoru; Ohno, Seiko; Hasegawa, Kanae; Makita, Naomasa; Nogami, Akihiko; Shimizu, Wataru; Aiba, Takeshi; Froguel, Philippe; Balkau, Beverley; Lantieri, Olivier; Torchio, Margherita; Wiese, Cornelia; Weber, David; Wolswinkel, Rianne; Coronel, Ruben; Boukens, Bas J; Bézieau, Stéphane; Charpentier, Eric; Chatel, Stéphanie; Despres, Aurore; Gros, Françoise; Kyndt, Florence; Lecointe, Simon; Lindenbaum, Pierre; Portero, Vincent; Violleau, Jade; Gessler, Manfred; Tan, Hanno L; Roden, Dan M; Christoffels, Vincent M; Marec, Hervé Le; Wilde, Arthur A; Probst, Vincent; Schott, Jean-Jacques; Dina, Christian; Redon, Richard. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death (англ.) // Nature Genetics : journal. — 2013. — ISSN 1061-4036. — doi:10.1038/ng.2712.
- ↑ Л. М. Макаров. Синдром Бругада. журнал «Лечащий врач».
Источник
Синдром Бругада – генетическое заболевание, которое обусловлено мутацией гена SCN5A, отвечающего за кодирование субъединиц (составляющих элементов) натриевых каналов мышечных клеток сердца (кардиомиоцитов).
Общая информация
Синдром Бругада был описан в 1992 году испано-бельгийскими кардиологами Педро и Хосепом Бругада. Это заболевание является причиной сердечной смерти в молодом возрасте в 50% случаев. Патологии больше подвержены мужчины.
Синдром Бругада очень распространен в государствах Юго-Восточной Азии – более 5 случаев на 10000 человек. В Европе и Северной Америке диагноз устанавливается реже – у 1 человека из 10000.
Причины
Этиология синдрома Бругада связана с мутацией гена SCN5A, который расположен на р плече третьей хромосомы. Кроме того, выделяют еще 4 гена, дефекты в которых могут привести к заболеванию. Все они отвечают за кодирование субъединиц натриевых каналов кардиомиоцитов, то есть белков, основной функцией которых является перемещение ионов натрия через мембрану мышечных клеток сердца.
Синдром Бругада передается по аутосомно-доминантному типу: для возникновения патологии у ребенка достаточно, чтобы он получил мутантный ген от одного из родителей.
Патогенез
Мутация гена SCN5A при синдроме Бругада приводит к инактивации (угнетению) натриевых каналов в кардиомиоцитах. В результате нарушается перемещение ионов натрия, которые играют значимую роль в электрохимических реакциях, приводящих к сокращению и расслаблению мышечных клеток сердца.
Как следствие, наблюдается аномальная электрическая активность сердечной мышцы (миокарда), и развивается пароксизмальная желудочковая тахикардия – резкое учащение сокращений до 150-180 ударов в минуту (норма – 60-80). Очаг возбуждения формируется в правом желудочке.
Пароксизмальная желудочковая тахикардия занимает первое место среди всех аритмий по степени угрозы для жизни. Без оказания помощи пациенту она способна перейти в фибрилляцию желудочков – состояние, которое сопровождается остановкой сердца.
Симптомы
Признаки синдрома Бругада на ЭКГ можно заметить с 5 лет. Манифестация симптомов происходит в возрасте 30-40 лет.
В зависимости от уровня изменений на ЭКГ при синдроме Бругада выделяют несколько клинико-электрографических типов. Полная форма включает следующие проявления:
- повышение сегмента ST над изолинией на 1 мм и выше в правых грудных отведениях, которое по форме напоминает очертания морды бультерьера (этот признак называют «типом бультерьера»);
- блокаду (полную или частичную) правой ножки пучка Гиса;
- периодическое увеличение интервала PR.
Основной симптом синдрома Бругада – приступы (пароксизмы) желудочковой тахикардии, которые обычно возникают вечером и ночью. Им могут предшествовать употребление алкоголя, нагрузка или лихорадка, связанная с инфекционным заболеванием. Иногда пароксизм начинается в состоянии полного покоя. Он сопровождается:
- ощутимыми толчками в области сердца и учащением сердцебиения;
- оглушенностью;
- потливостью;
- головокружением;
- появлением «мушек» перед глазами.
Многие пациенты теряют сознание (возникает синкопе). В 89% случаев через 20-30 секунд состояние нормализуется. У остальных происходит остановка сердца из-за фибрилляции желудочков.
Диагностика
Диагностика синдрома Бругада проводится с помощью ЭКГ. При необходимости выполняется электрокардиография с высокими грудными отведениями (электроды накладываются выше, чем обычно), а также регистрация кардиограммы после предварительной стимуляции сердца. Целесообразно осуществление суточного мониторинга ЭКГ.
Для постановки диагноза у бессимптомных пациентов может проводиться фармакологическая проба: введение блокаторов натриевых каналов (аймалина, прокаинамида, флекаина) с последующей ЭКГ. При наличии синдрома Бругада лекарства провоцируют пароксизм желудочковой тахикардии. Этот тест делается только в условиях больницы. Дополнительно могут назначаться МРТ мозга и нейросонография.
Подтвердить синдром Бругада можно с помощью генетического исследования, в ходе которого обнаруживается мутировавший ген. Анализ рекомендуется проводить не только пациентам, но и их родственникам. Его точность – 20-30%.
Синдром Бругада дифференцируют от острого перикардита, гиперкалиемии, инфаркта миокарда, дисплазии правого желудочка, полиморфной желудочковой тахикардии и так далее.
Лечение
Эффективное медикаментозное лечение синдрома Бругада не разработано. Для предупреждения пароксизмов желудочковой тахикардии используются антиаритмические препараты – хинидин, дизопирамид, амиодарон, пропранолол. Введение блокаторов натриевых каналов противопоказано.
Наиболее действенным методом уменьшения риска внезапной сердечной смерти при синдроме Бругада является хирургическая операция, в ходе которой устанавливается кардиовертер-дефибриллятор. Этот кардиостимулятор контролирует сердечный ритм и выполняет дефибрилляцию с помощью внутрисердечных электродов в случае приступа желудочковой аритмии.
Установка кардиовертера-дефибриллятора проводится пациентам с высокой вероятностью сердечной смерти. Факторы риска:
- случаи внезапной сердечной смерти в семейном анамнезе;
- синкопальные состояния;
- беспричинные изменения ЭКГ;
- подтвержденная мутация гена SCN5A.
Прогноз
Прогноз при синдроме Бругада неблагоприятный – 11% пациентов погибают в молодом возрасте от внезапной сердечной смерти.
Установка кардиовертера-дефибриллятора положительно сказывается на качестве жизни пациентов. Им необходимо ежегодно посещать кардиохирурга, а также менять устройство раз в 4-6 лет.
Людям с синдромом Бурагада следует вести здоровый образ жизни, сбалансировано питаться, исключить стрессы и отказаться от экстремальных занятий. Эти мероприятия помогают снизить вероятность приступов тахикардии.
Профилактика
Синдром Бругада имеет генетическую природу, что обуславливает невозможность разработки мер по его профилактике. Парам с отягощенным семейным анамнезом следует консультироваться с генетиком при планировании беременности.
Источник