Синдром блоха сульцбергера мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
Названия
Название: Синдром Блоха-Сульцбергера.

Синдром Блоха-Сульцбергера
Описание
Синдром Блоха. Сульцбергера — наследственная форма нарушения пигментации кожи, которая часто сочетается с пороками развития зубов, волос, ногтей и глаз. Симптомы заболевания характеризуются выраженной стадийностью – сначала на коже появляется эритематозная сыпь в виде пятен и линий, затем на ее месте развивается гиперкератоз, сменяющийся пятнами и последующей гипопигментацией с атрофией кожных покровов. Диагностика синдрома Блоха-Сульцбергера производится на основании данных настоящего статуса больного, гистологического исследования образцов кожи в области поражения, изучения наследственного анамнеза и молекулярно-генетических анализов. Специфического лечения этой патологии на сегодняшний момент не существует, используют симптоматические и поддерживающие мероприятия различного характера.
Дополнительные факты
Синдром Блоха-Сульцбергера (семейная форма недержания пигмента, нейрокожный меланобластоз) – генетическое заболевание, характеризующееся нарушением метаболизма меланина в коже и рядом сопутствующих пороков развития. Впервые это заболевание было описано в 1926 году швейцарским дерматологом Б. Блохом, затем более детальное изучение данной патологии провел американский педиатр М. Сульцбергер в 1929 году и, независимо от предыдущих исследователей, немецкий врач Г. Сименс. Именно поэтому в литературе можно найти другое название этого заболевания – синдром Блоха-Сименса. Удалось выяснить, что патология наследуется сцепленно с Х-хромосомой, при этом мутантный аллель является доминантным. По этой причине синдром Блоха-Сульцбергера во много раз чаще встречается у девочек – половое распределение составляет примерно 6:210, так как наличие этой мутации у эмбрионов мужского пола практически всегда является летальным и приводит к самопроизвольному прерыванию беременности. Развитие заболевания у мальчиков может быть обусловлено генетическим мозаицизмом, наличием сопутствующего синдрома Клайнфельтера или редких точечных «мягких» мутаций. Общая встречаемость синдрома Блоха-Сульцбергера составляет примерно 1 случай на 75 000 новорожденных.
Синдром Блоха-Сульцбергера
Причины
При синдроме Блоха-Сульцбергера происходит повреждение гена IKBKG, который располагается на Х-хромосоме. Продуктом его экспрессии является многофункциональный сложный белок – регуляторная субъединица NEMO-ингибиторной киназы, участвующей в сигнальной системе важного транскрипционного фактора (NF-каппа-B). Этот фактор и соответствующий ему сигнальный путь регулирует огромное количество различных процессов в организме человека – участвует в процессах адаптации при стрессе, иммунном ответе, некоторых формах воспалительных реакций, процессах клеточной адгезии, а также тормозит процессы апоптоза. Наиболее часто причиной синдрома Блоха-Сульцбергера становятся крупные транслокации и делеции гена IKBKG, в результате чего экспрессия и выделение белка с этого гена полностью прекращаются.
Поскольку у женщин имеется две Х-хромосомы, при наличии второй нормальной аллели гена IKBKG такая мутация не угрожает жизни, но обуславливает развитие синдрома Блоха-Сульцбергера. Известен факт, что в соматических клетках женского организма всегда активна только одна Х-хромосома, тогда как вторая сконденсирована в половой хроматин. Значительная вариабельность выраженности симптомов заболевания обусловлена распределением клеток, где активна хромосома именно с мутантной формой гена IKBKG. Как следствие, в вышеуказанных клетках не образуется регуляторной субъединицы NEMO-ингибиторной киназы, что и приводит к характерным порокам развития, формирующим клиническую картину синдрома Блоха-Сульцбергера. Кожные симптомы связаны с нарушением проницаемости мембран меланоцитов (в результате чего практически весь пигмент беспрепятственно покидает клетки) и аутоиммунными реакциями.
В отличие от женщин, у мужчин в норме есть лишь одна Х-хромосома, поэтому при наличии нонсенс-мутации в гене IKBKG выделение важного белка не происходит абсолютно во всех клетках организма. Это становится причиной массированного апоптоза гепатоцитов еще на этапе внутриутробного развития – в норме этот процесс задерживается как раз системой NF-каппа-B. Развитие нарушений, подобных синдрому Блоха-Сульцбергера, у мальчиков возможно при наличии сопутствующего синдрома Клайнфельтера (кариотипа XXY) или генетического мозаицизма, когда только часть клеток в организме имеет дефект гена IKBKG. В последние годы были выявлены точечные мутации этого гена, которые не приводят к полной остановке транскрипции, но изменяют структуру конечного белка. Однако чаще всего у мальчиков с такими дефектами возникает не синдром Блоха-Сульцбергера, а другие генетические заболевания – эктодермальные дисплазии, иммунодефициты, пороки развития скелета.
Симптомы
Одним из наиболее выраженных и распространенных проявлений синдрома Блоха-Сульцбергера является дерматоз, который обнаруживается при рождении или (реже) возникает на протяжении первых дней жизни новорожденного. В развитии изменений кожных покровов при этой патологии наблюдается характерная стадийность, что также является важным диагностическим признаком. Локализация таких изменений – на боковых поверхностях конечностей, туловища, шеи, вдоль линий Шарко или проекций основных нервных стволов. В большинстве случаев выделяется четыре основных стадии кожных симптомов синдрома Блоха-Сульцбергера:
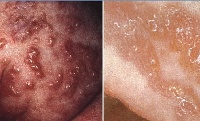
1. Я стадия — воспалительная или везикулобуллезная. Начинается при рождении больного или на протяжении 2-3 недель жизни и длится до возраста 3-8 месяцев. На этом этапе заболевания возникают везикулы, эритематозная сыпь, возможно развитие пузырей и пустул. С учетом возраста больных синдромом Блоха-Сульцбергера существует определенный риск инфекционных осложнений на пораженных участках кожи.
2. Я стадия — гипертрофическая или веррукозная. Характеризуется развитием на пораженных участках тела гиперкератоза в виде бляшек, бородавчатых и лихеноидных разрастаний. Их распределение, как правило, симметричное и линейное, вдоль линий Шарко или проекций нервных стволов. Длительность этой стадии синдрома Блоха-Сульцбергера составляет несколько месяцев (до возраста одного года), у некоторых больных может отсутствовать.
3. Я стадия — пигментная. На этом этапе заболевания у больных на пораженных участках кожи возникают очаги гиперпигментации различных форм и размеров темно-коричневого цвета. Почти у половины пациентов с синдромом Блоха-Сульцбергера такие очаги появляются на неизмененных участках тела и не связаны с высыпаниями, характерными для предыдущих стадий. Длительность гиперпигментации составляет несколько лет, обычно – до периода полового созревания.
4. Я стадия — атрофическая. Характеризуется потерей пигмента на очагах поражения с развитием признаков атрофии кожи. У некоторых больных синдромом Блоха-Сульцбергера такие проявления могут быть выражены очень слабо, в отдельных случаях симптомы заболевания полностью исчезают после завершения полового созревания.
Диагностика
Для определения синдрома Блоха-Сульцбергера используют множество диагностических методов и техник – дерматологический осмотр, изучение наследственного анамнеза, гистологическое исследование пораженных участков кожных покровов, молекулярно-генетические анализы. При осмотре выявляются разнообразные (в зависимости от возраста больных и стадии заболевания) изменения кожи эритематозного, везикулярного или гиперкератического характера, у старших пациентов может определяться очаговая гипер- или гипопигментация кожи. Помимо этих проявлений, при синдроме Блоха-Сульцбергера возможна дистрофия ногтей, алопеция, аномалии строения зубов.
Наследственный анамнез может выявить семейный, доминантный и сцепленный с Х-хромосомой характер наследования патологии. В некоторых случаях у матери больной в анамнезе отмечается несколько случаев самопроизвольного прерывания беременности – это связано с внутриутробной смертью плода мужского пола. Результаты гистологического исследования тканей кожи при синдроме Блоха-Сульцбергера зависят от стадии заболевания – на первом этапе обнаруживается спонгиоз, развитие эпидермальных пузырей, заполненных эозинофилами и фибриновыми массами. На второй стадии патологии выявляются признаки внутриэпителиальной кератинизации, акантоз и гиперкератоз, в дерме отмечается отек с нейтрофильной и эозинофильной инфильтрацией. На третьей стадии синдрома Блоха-Сульцбергера воспалительные изменения в дерме (отек, инфильтрация) исчезают, но наблюдается значительное накопление пигмента в верхних слоях кожи. Четвертая стадия характеризуется исчезновением пигмента, развитием фиброзной ткани и частичным исчезновением придатков кожи.
Молекулярно-генетическая диагностика синдрома Блоха-Сульцбергера выполняется врачом-генетиком и может быть произведена несколькими основными техниками. Прямое автоматическое секвенирование последовательности гена IKBKG позволяет выявить практически любые изменения в его структуре. Транслокации и делеции значительных участков гена, часто выступающие в качестве причины синдрома Блоха-Сульцбергера, можно обнаружить при помощи методики FISH-анализа. Данное заболевание также может быть подтверждено посредством исследования инактивации Х-хромосом в клетках пораженных тканей.
Лечение
Специфического лечения синдрома Блоха-Сульцбергера на сегодняшний момент не существует, кожные поражения на воспалительном этапе заболевания обрабатываются антисептическими средствами и растворами для предотвращения инфекционных осложнений. Кроме того, рекомендуется местное назначение глюкокортикоидных стероидов для уменьшения воспаления, однако такое лечение следует производить с осторожностью, учитывая высокую проницаемость кожных покровов у детей младшего возраста. Другие проявления синдрома Блоха-Сульцбергера (пороки развития зубов, глаз) лечат при наличии показаний.
Источник
Рубрика МКБ-10: Q82.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q82 Другие врожденные аномалии пороки развития кожи
Определение и общие сведения[править]
Недержание пигмента
Синонимы: Incontinentia pigmenti, синдром Блоха-Сульцбергера, синдром Блоха-Сименса
Системное заболевание, проявляющееся своеобразными стадийными изменениями кожи в сочетании с патологией глаз, зубов, волос и ногтей, ЦНС и костно-мышечного аппарата.
Эпидемиология
Наследование носит Х-сцепленный доминантный характер. Подавляющее большинство случаев семейные.
Распространенность составляет приблизительно 1 / 143,000. Соотношение женщин и мужчин составляет 20: 1.
Этиология и патогенез[править]
Недержание пигмента вызывается семейной (10-25%) или спорадической de novo (> 50%) мутацией NF-kappaB естественного модулятора гена IKBKG (ранее NEMO). 4-10 делеции экзона лежат в основе 80% случаев заболевания.
Клинические проявления[править]
Заболевание существует с рождения или проявляется в первые недели жизни. Характерна стадийность кожных изменений.
• Стадия I (пузырьковая, или воспалительная). Она возникает в первые часы жизни в виде отечной эритемы с везикулобуллезными, реже уртикарными элементами, которые склонны к линейному расположению. Содержимое пузырьков прозрачное, в нем обнаруживают эозинофилы. При вскрытии и подсыхании пузырьков образуются мелкие эрозии и корочки. Общее состояние ребенка обычно не нарушено, в периферической крови — эозинофилия.
• Стадия II (веррукозная). Она развивается приблизительно через 2-3 мес и характеризуется лентикулярными ороговевающими папулами, расположенными преимущественно линейно в зоне бывших везикул или беспорядочно, напоминая бородавчатый невус. Веррукозные изменения кожи сохраняются в течение нескольких месяцев. На ладонях и подошвах может быть диффузный гиперкератоз.
• Стадия III (гиперпигментация). Она наступает через 3-6 мес от начала заболевания. На месте регрессировавших очагов развивается пигментация коричневатого или темно-серого цвета, напоминающая брызги грязи на светлом фоне, в виде полосок и завихрений.
• Стадия IV (гипопигментация). Она проявляется на 2-3-м десятилетии жизни. Пигментация у части больных сменяется депигментацией, умеренно выраженной атрофией, очаговым склерозом.
Стадийность наблюдают не всегда. Более 10% больных имеют только пигментацию. Может быть только изолированная воспалительная стадия. Признаки различных стадий могут существовать одновременно.
Из внекожных изменений наиболее часто возникают нарушения со стороны нервной системы и органа зрения.
Недержание пигмента (incontinentia pigmenti): Диагностика[править]
Типичные поражения кожи и генетическое тестирование являются достаточными для установления диагноза.
Наблюдается также лейкоцитоз и эозинофилия. Гистологические изменения зависят от стадии заболевания.
Дифференциальный диагноз[править]
Этап I может быть неправильно диагностирован как буллезный импетиго, врожденный буллезный эпидермолиз, герпес или ветряную оспу.
Дифференциальная диагностика II стадии включает в себя бородавки, контагиозный моллюск и синдром эпидермального невуса.
Любое состояние с линейной и окружной пигментацией пересекается с клиникой стадии III.
Стадия IV напоминает рубцевание, витилиго, гипомеланоз Ито или другие гипопигментозы с локализованной алопецией.
Дополнительно следует рассматривать синдром Нейджели-Франческетти-Ядассона и болезнь Норри.
Недержание пигмента (incontinentia pigmenti): Лечение[править]
Лечение симптоматическое. На ранних стадиях назначают 10% мазь цинка оксида, мази с глюкокортикоидами, анилиновые красители, при инфицировании — антибиотики. При развитии массивных веррукозных изменений используют изотретиноин.
Прогноз
Ожидаемая продолжительность жизни в норме. Лица, не имеющие аномалий ЦНС, как правило, имеют нормальное физическое и умственное развитие.
Профилактика[править]
Недержание пигмента наследуется Х-сцепленно доминантно. Заболевшая женщина имеет 50% риск передачи патологии потомству. Больные мужского пола должны быть проверены на 47, XXY кариотип.
Прочее[править]
Источники (ссылки)[править]
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
В 95% случаев встречается у женщин.Наследование носит Х-сцепленный доминантный характер.Подавляющее большинство случаев семейные.Примерно в 80% случаев новых мутаций установлено их отцовское происхождение.
Причина — локус патологического гена — Xq28.Мутантный ген считают летальным для гомозиготных плодов мужского пола.У плодов женского пола к моменту рождения происходит селективная элиминация клеток с экспрессией патологического гена,поэтому у новорожденных девочек имеет место крайне неравномерная
инактивация
Х-хромосомы в данном локусе (в кератиноцитах,фибробластах,лейкоцитах периферической крови).Основным нарушением считают нестабильность хромосом.
Также придают значение нарушению иммунной толерантности,в связи с чем происходит аутоиммунная атака на клоны клеток эктодермального происхождения,имеющие аномальные поверхностные антигены,или происходит преждевременная запрограммированная гибель клонов измененных клеток.
Заболевание существует с рождения или проявляется в первые недели жизни.Характерна стадийность кожных изменений.
Первая стадия (везикулезно-буллезная,или воспалительная)
 Возникает уже в первые часы жизни,иногда с момента рождения,в виде отечной эритемы с везикуло-буллезными,реже уртикарными,элементами,которые склонны к линейному расположению.Содержимое пузырьков обычно прозрачное,при их вскрытии и подсыхании образуются мелкие эрозии и корочки.Высыпания возникают приступами,распространяясь
Возникает уже в первые часы жизни,иногда с момента рождения,в виде отечной эритемы с везикуло-буллезными,реже уртикарными,элементами,которые склонны к линейному расположению.Содержимое пузырьков обычно прозрачное,при их вскрытии и подсыхании образуются мелкие эрозии и корочки.Высыпания возникают приступами,распространяясь
на новые участки
кожи.Наиболее часто они расположены на конечностях.В содержимом везикуло-буллезных
элементов
обнаруживают
эозинофилы,в периферической крови — эозинофилия.Общее состояние ребенка обычно не нарушено
Вторая стадия (веррукозная)
 Развивается приблизительно через 2–3 мес и характеризуется лентикулярными ороговевающими папулами,расположенными преимущественно линейно в зоне бывших везикул или беспорядочно,часто напоминая бородавчатый невус.Веррукозные изменения кожи сохраняются в течение нескольких месяцев.На ладонях и подошвах может быть диффузный гиперкератоз.
Развивается приблизительно через 2–3 мес и характеризуется лентикулярными ороговевающими папулами,расположенными преимущественно линейно в зоне бывших везикул или беспорядочно,часто напоминая бородавчатый невус.Веррукозные изменения кожи сохраняются в течение нескольких месяцев.На ладонях и подошвах может быть диффузный гиперкератоз.
Третья стадия (гиперпигментация)
 Наступает через 3–6 мес от начала заболевания.На месте регрессировавших очагов развивается пигментация коричневатого или темно-серого цвета,напоминающая «брызги грязи» на светлом фоне,в виде полосок и завихрений,ветвистого,звездчатого узора или мраморного торта.
Наступает через 3–6 мес от начала заболевания.На месте регрессировавших очагов развивается пигментация коричневатого или темно-серого цвета,напоминающая «брызги грязи» на светлом фоне,в виде полосок и завихрений,ветвистого,звездчатого узора или мраморного торта.
Четвертая стадия (гипопигментация и атрофия)
Проявляется на 2–3-м десятилетии жизни.Пигментация у части больных сменяется депигментацией,умеренно выраженной атрофией,очаговым склерозом.
Стадийность наблюдают не всегда.Более 10% больных имеют только пигментацию.Может быть только изолированная воспалительная стадия.Признаки различных стадий могут существовать одновременно.Описаны случаи с отсроченной «реактивацией» воспалительных и буллезных высыпаний в области линейных очагов гиперпигментации спустя месяцы и годы после их заживления.Эти случаи по времени совпадают с перенесенной респираторно-вирусной или бактериальной инфекцией,сопровождающейся лихорадочным состоянием.
Волосы обычно не изменены,но приблизительно в 25% случаев выявляются участки атрофической алопеции,развивающейся в детском возрасте.Ногтевые пластинки обычно маленькие и слегка дистрофичные.
Атипичные формы:сетчатый пигментный дерматоз,синдром Асбо–Хансена,недержание пигмента ахромичное Ито.
Внекожные изменения со стороны:
- нервной системы (припадки,умственная отсталость,спастические параличи,гидроцефалия и др.)
- глаз (страбизм,нистагм,гипертелоризм,эпикант,микрофтальмия,катаракта,голубые склеры,псевдоглиома,пигментация,отслойка сетчатки,атрофия зрительного нерва,слепота).
- зубов (задержка смена молочных зубов на постоянные,частичном или полном их отсутствии,коническая форма зубов)
Диагноз основывают на особенностях клинической картины,данных гистологического исследования,результатах обследования у невропатолога,ортопеда,окулиста,стоматолога и других специалистов.
Необходим тщательный осмотр родственников больного для обнаружения малых признаков заболевания.Зубные аномалии могут быть его единственным проявлением,как и депигментированные сетчатые очаги.
Гистологические изменения зависят от стадии заболевания.Вначале выявляют эозинофильный спонгиоз: внутрипидермальные пузыри
с выраженным экзоцитозом эозинофилов в их полость и окружающий эпидермис.На стадии кератотических изменений выявляют гиперкератоз,акантоз,неравномерный папилломатоз и многочисленные дискератотические клетки.На стадии гиперпигментации преобладают явления недержания пигмента.Характерно проникновение пигмента в дерму и накопление его в меланофагах.На стадии гипопигментации количество
пигмента в эпидермисе снижено.
Воспалительная стадия:
- Буллезный эпидермолиз
- Эпидемическая пузырчатка новорожденных
- Неонатальный герпес
- Сифилитическая пузырчатка
Веррукозная стадия:
- Линейный эпидермальный невус
- Себорейный невус
- Полосовидный(линейный) лишай
- Линейная форма болезни Дарье
- Линейная форма красного плоского лишая
Пигментная стадия:
- Гипермеланоз невоидный линейный и вихревидный
- Фокальная дермальная гипоплазия Гольтца
- Синдром Олбрайта
- Гипомеланоз Ито
Лечение симптоматическое.На ранних стадиях назначают 10% мазь цинка оксида,мази с глюкокортикоидами,анилиновые красители,при инфицировании — антибиотики.При развитии массивных веррукозных изменений используют изотретиноин.Показано наблюдение невропатолога,ортопеда,стоматолога и других специалистов.Рекомендовано медико-генетическое консультирование
Источник