Синдром арта что это такое

Мы изначально подозревали у Егора генетику, и поэтому даже после того, как генетики из Московского Генетического Центра (МГЦ) отправили нас два раза (в 2013 и 2014) домой, мол, у вас нет ничего генетического, занимайтесь неврологией, мы все равно не сдались и продолжили копать эту тему. В конце концов решили сделать Егору секвенирование экзома, когда как раз этот анализ упал в цене. В итоге ответ был найден — синдром Арта.
Синдром Арта
Описание
Описание взято отсюда и переведено мною.
К развитию синдрома Арта приводят мутации в гене PRPS1, расположенном на длинном плече Х-хромосомы. Ген кодирует белок, который катализирует фосфорибозилирование рибозы-5-фосфат до 5-фосфорибозил-1-пирофосфата, что необходимо для метаболизма пурина и биосинтеза нуклеотидов.
Мальчики с синдромом Арта имеют нейросенсорную тугоухость, при которой происходит полная или почти полная потеря слуха. Другие особенности расстройства включают слабый мышечный тонус (гипотония), нарушение координации (атаксия), задержку развития и умственную отсталость. В раннем детстве развивается потеря зрения, вызванная дегенерацией нервов, которые несут информацию от глаз к мозгу (атрофия зрительного нерва). Также дети испытывают потерю чувствительности и слабость в конечностях (периферическая невропатия).
При синдроме Арта характерна восприимчивость к инфекциям, особенно верхних дыхательных путей. Инфекции и связанные с ними осложнения, могут приводить к смерти в раннем детстве.
Полезная информация по синдрому
Не скажу, что я много нарыл информации, да и пригодится она очень малому количеству людей, но тем не менее выложу сюда основное. Всяко больше пользы, чем она у меня на компе хранится будет.
https://www.ncbi.nlm.nih.gov/books/NBK2591/ — подробное описание синдрома.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-9-24 — статья про сидром Арта и похожие заболевания.
https://www.ncbi.nlm.nih.gov/pubmed/?term=Prps1 — все статьи на Пабмеде по гену PRPS1. Можно вбить в поисковую строчку и другой ген, если надо. Так вот если заходить во все статьи из списка и жать на Author Information, то увидите емейлы авторов. Можно написать им всем, говорят пара человек может согласится работать бесплатно с мутацией. Как раз этим я сейчас и занимаюсь.
PRS1_1.pdf — ссылка на скачивание статьи от Dr.Arjan P.M. de Brouwer, он профессор из Нидерландов. Я ему писал и он сначала вроде откликнулся, да-да, посмотрит анализы, свяжет с коллегами и все такое, но потом пропал. Спустя пару месяцев я ему еще раз написал, но уже без ответа.
https://informahealthcare.com/doi/abs/10.3109/14992027.2012.736032 — статья от универсистета в Майми, им я тоже писал, но безрезультатно.
Мутации в гене PRPS1 приводят не только к синдрому Арта, но и к Шарко-Мари-Тута, суперактивность фосфорибозилпирофосфат синтетазы. У каждого синдрома есть OMIM номер (он будет в заключении о секвенировании, пример ниже), который можно ввести вот здесь (откроется для синдрома Арта), и тоже получим некоторую информацию по синдрому.
Егор
Мутация у Егора
Вырезка из заключения секвенирования (полный вариант на русском, на английском):
Выявлена ранее не зарегистрированная гемизиготная миссенс мутация в экзоне 2 гена PRPS1 (chrX:106882652; C>T) что приводит к замене аминокислоты триптофана на аргинин в кодоне 84 (p.R84W; ENST00000372435). Гомозиготные мутации в гене PRPS1 приводят к Х- сцепленным рецессивным синдромам, таким как: Arts syndrome (OMIM#301835), Х-сцепленный рецессивный тип Шарко-Мари-Тута (OMIM#311070) и суперактивность фосфорибозилпирофосфат синтетазы I (OMIM#300661). Мутация p.R84W не зарегистрирована в базах данных 1000 геномов, а в базах данных SIFT, LRT and Mutation Taster расценивается как вероятно патогенная.
Несмотря на то, что мутация не совсем изучена, она находится в том же самом гене PRPS1. Также генетик Дадали на последнем приеме (из-за которого я и остался в Москве) мне сказала, что она специально посмотрела эту мутацию в базе и считает ее не вероятно-патогенной, а именно патогенной. В любом случае клиническая картина Егора полностью соответствует описанию синдрома Арта, и это как раз и говорит о данном заболевании. Генетик Солониченко сказал примерно тоже самое.
К сожалению, это именно генетика, и мутация спонтанная (так называемый гонадный мозаицизм), и невозможно установить, почему так случилось. Синдром Арта может передаваться только по материнской линии, но у Дарьи все чисто, мы специально проверили. И специально проверили PRPS1 у Егора еще раз (результат). Насколько я понял, так чаще всего и бывает, когда заболевания не наследственные, а на пустом месте возникают у детей от здоровых родителей. Кстати, это говорит и о том, что если заводить еще детей, то рисков нет никаких…

Окончательный диагноз — синдром Арта
Для меня загадка, почему генетики с самого начала не предположили этот синдром, который сразу все эти непохожие друг на друга симптомы собрал в себе. Скорее всего, потому что Арт очень редкий синдром, вероятность заболевания которого чуть ли не 1 на миллион, и та же Дадади за всю свою долгую практику видела таких людей буквально одного. С другой стороны, почему-то было проще нас послать искать проблемы в неврологии, чем задуматься и попробовать объяснить наши нестыкующиеся симптомы. Никогда не забуду фразу Галкиной, у которой мы были в МГЦ на приеме — «Понимаете, тут работать надо, а вас много очень». И дело, конечно же, не в Галкиной, а в системе здравоохранения, раз она не может обеспечить врачей временем, но слышать от нее это было несколько странно. Только благодаря нашей интуиции, мы и сидром этот нашли, и вообще многое делали правильно.

Прогнозы
Прогнозы… Да, какие тут могут быть прогнозы. Если официально, то все не очень хорошо. А если неофициально, то кто его знает, как и что будет. Пока есть надежда, нельзя опускать руки. У нас есть отличный пример перед глазами (https://antysk.livejournal.com/), когда ребенок согласно диагнозу не должен ни ходить, ни общаться, овощем быть, ан нет, ходит и общается. Так что, откуда мы знаем, как собиралась статистика по детям с синдром Арта, может ими вообще никто не занимался и родители совсем в процесс не включались. А ведь даже легкую умственную отсталость можно так развить у ребенка, что он почти от обычного человека отличаться не будет.

Когда-то все только начиналось

Когда по комнате бегает вот такая забавная шляпа, то уже не так страшно все кажется
Так что все выше написанное, не означает, что мы опустим руки. Реабилитировать все равно будем продолжать, и никак иначе, тем более есть результаты! Сейчас вот в Китае, а до этого были во Фрязино и Олинеке, и видим, что Егор развивается, пусть и медленно.
P.S. Честно говоря, мы еще не до конца пришли в себя, надо как-то все это пережить и принять. Вроде, кажется, приняли глухоту, потом стало ясно, что не ходит, теперь вот эта генетика. И каждый раз, думаешь, ох, вот дураки из-за какой-то глухоты переживали, нам бы ее сейчас вместо всего этого набора…
Как мы экономим на добавках и витаминах
Витамины, пробиотики, муку без глютена, косметику, спортивное питание, мы заказываем на iHerb.com (по ссылке скидка 5$). Доставка в Москву всего 1-2 недели. Многое дешевле в несколько раз, нежели брать в российском магазине.
Получить скидку 5$ →
Источник
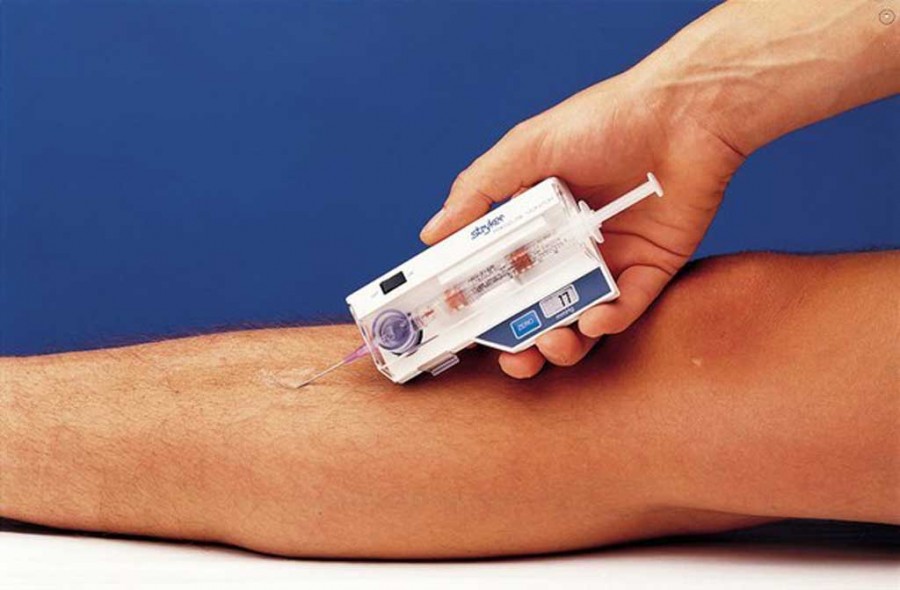
Компартмент-синдром, называемый также синдромом сдавливания, возникает в тех случаях, когда в замкнутом мышечном пространстве нарастает избыточное давление. Обычно этот синдром появляется в результате кровотечения или опухания после травмы. Высокое давление при компартмент-синдроме очень опасно, так как оно затрудняет приток крови к поврежденным тканям и отток ее от них. В экстренных случаях для предупреждения перманентного повреждения органов или тканей бывает необходимо хирургическое вмешательство.
КАК РАЗВИВАЕТСЯ КОМПАРТМЕНТ-СИНДРОМ
Чаще всего компартмент-синдром возникает в ногах, руках и брюшной полости — в группах органов или мышц, объединенных в общей анатомической полости, называемой компартментом. Стенки компартментов формируются из нерастяжимого переплетения волокон соединительной ткани — фасций. В результате травмы в компартменте может скапливаться кровь или эдема — жидкость, вырабатывающаяся в результате воспаления или травмы. Из-за того что стенки фасции не могут существенно растягиваться, в компартменте повышается давление, что затрудняет кровоток внутри поврежденной анатомической полости. В результате может возникнуть тяжелое повреждение тканей, приводящее к утрате некоторых функций организма и даже к фатальному исходу.
ПРИЧИНЫ РАЗВИТИЯ КОМПАРТМЕНТ-СИНДРОМА
Острый компартмент-синдром — наиболее часто встречающаяся форма этого заболевания. В 75% случаев он является следствием перелома рук или ног и развивается достаточно быстро — в течение часов или дней после травмы.
Синдром может развиться как непосредственно от перелома (из-за повышенного давления от кровотечения или накопления жидкости), так и на более позднем этапе — в результате наложения гипса или проведения хирургической операции.
Причиной острого компартмент-синдрома может стать не только перелом костей, но и другие травмы, в том числе:
различные травмы при транспортных авариях (автомобильных, авиационных и прочих),
ожоги,
слишком тугие перевязки,
продолжительное сдавливание конечности в период потери сознания,
операция на кровеносных сосудах рук или ног,
тромб в кровеносных сосудах рук или ног,
слишком энергичные физические упражнения — особенно при сочетании растяжек с большой внешней нагрузкой.
Прием анаболических стероидов также содействует развитию компартмент-синдрома.
Другая форма компартмент-синдрома — хронический компартмент-синдром, называемый также компартментальным синдромом физической нагрузки,— развивается в течение нескольких дней или недель. Его причиной могут стать регулярные физические нагрузки большой интенсивности. Обычно это происходит в нижней части ног, ягодицах или бедрах.
Симптомами хронического компартментального синдрома являются ноющая или судорожная боль в поврежденных конечностях, ограниченность их функционирования, слабость, онемение или покалывание. В тяжелых случаях могут быть повреждены нервы, что приводит к свисанию стопы. Иногда возникает мышечная грыжа, сопровождаемая отеком и опуханием.
Компартмент-синдром брюшной полости почти всегда развивается в результате тяжелой травмы, особенно сопровождаемой шоком, перелома таза, ожога, хирургической операции, часто при пересадке печени, сепсиса или во время тяжелой болезни. Находящиеся в брюшной полости органы — печень, желудок, почки и другие — могут быть серьезно, а иногда и необратимо повреждены.
ЛЕЧЕНИЕ КОМПАРТМЕНТАЛЬНОГО СИНДРОМА
В первую очередь надо постараться предотвратить компартментальный синдром. При серьезных травмах рук или ног, требующих наложения гипсовых повязок или шины, необходимо держать конечности высоко поднятыми вверх и охлаждать их льдом, что уменьшит опухание. Острый компартмент-синдром лечится с помощью хирургической операции (фасциотомии).
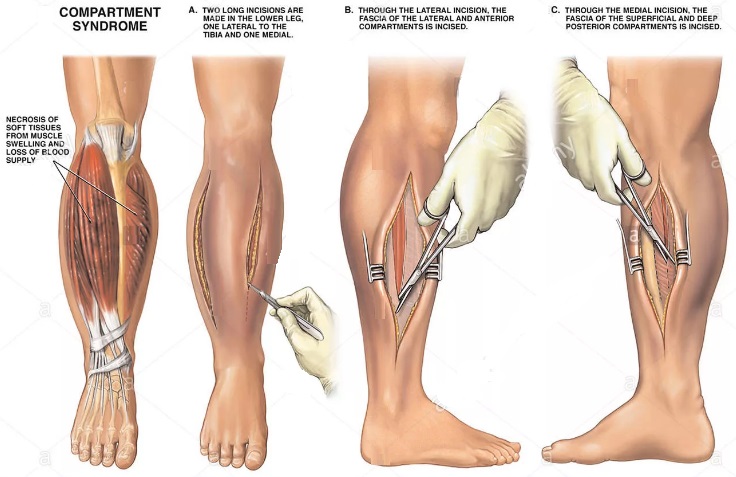
В ходе операции толстые фиброзные связки, устилающие мышцы, разделяются, что позволяет мышцам расшириться и понизить давление в компартменте. Для ускорения заживления раны после операции используют гиперборическую кислородную камеру.
Журнал Медицинский офис №217
Автор: Яков Грин
Фото: Яндекс Картинки
Источник
Синдром Картагенера – это генетическая патология цилиарного аппарата, ведущая к развитию хронических риносинуситов, бронхитов, бронхоэктазов, сочетающаяся с обратным расположением органокомплекса «сердце–лёгкие». Заболевание дебютирует в младенческом возрасте и характеризуется частыми гнойно-воспалительными процессами верхних и нижних дыхательных путей. Диагностируется с помощью лучевых методов исследования органов грудной клетки, биопсии слизистых оболочек бронхов или носа. В консервативной терапии используются антибиотики, кортикостероиды, бронхолитики. При необходимости выполняются хирургические операции в области назальных синусов, частичная резекция лёгких.
Общие сведения
Синдром Картагенера (триада Зиверта-Картагенера, синдром неподвижных ресничек) относится к наследственным болезням из группы первичных цилиарных дискинезий. Одним из первых в 1902 году заболевание описал российский врач А.К. Зиверт. В 1933 году швейцарский терапевт М. Картагенер детально изучил триаду и доказал её наследственную природу. Синдром Зиверта-Картагенера является редкой генетической патологией и встречается у 1 новорождённого на 25 000 – 50 000 родившихся живыми детей. У 50% пациентов с данным пороком встречается полная транспозиция (зеркальное расположение) внутренних органов. Патология нередко сочетается с другими врождёнными аномалиями (полидактилия, «заячья губа», глухонемота и прочие).

Синдром Картагенера
Причины
Причиной возникновения триады Картагенера являются мутации генов, отвечающих за нормальное функционирование ресничек и жгутиков различных клеток человеческого организма. Генетические дефекты передаются по аутосомно-рецессивному типу наследования. Первичная цилиарная дискинезия наблюдается у нескольких членов одной семьи. Заболевание проявляется у половины носителей мутантных генов. У родственников больного могут отсутствовать один или несколько признаков классической триады.
Патогенез
Из-за генетического дефекта нарушается синтез структурных белков жгутиков и ресничек. Цилиарный аппарат больного неподвижен или колеблется асинхронно. Во внутриутробном периоде из-за неправильного движения реснитчатого эпителия эмбриона должным образом не выполняется поворот внутренних органов, что приводит к их полному или частичному обратному расположению.
Неспособность мерцательного эпителия дыхательных путей к синхронному движению резко снижает дренажную функцию респираторной системы. Мокрота застаивается. При присоединении вторичной инфекции легко возникают очаги воспаления, формируются бронхоэктазы. Неподвижность или аномальное колебание ресничек эпителия, выстилающего придаточные пазухи носа и евстахиеву трубу, провоцирует рецидивирующие синуситы, евстахииты и отиты. Отсутствие или дисфункция жгутиков сперматозоидов затрудняет их передвижение и является причиной снижения способности к оплодотворению у мужчин.
Симптомы
С первых месяцев жизни у детей с синдромом Картагенера неподвижных ресничек возникают частые рецидивирующие эпизоды насморка и кашля, сопровождающиеся подъёмом температуры до фебрильных цифр. Выделения из носа обычно бывают гнойного характера. Нередко к явлениям ринита присоединяются признаки евстахиита и отита. Дети испытывают головные боли распирающего характера, пульсирующую «стреляющую» боль в ушах.
К 2-3 летнему возрасту у ребёнка происходит хронизация бронхита, кашель становится постоянным. Отделяется слизисто-гнойная (жёлто-зелёная) мокрота. Присоединяется синдром обструкции верхних дыхательных путей. Больного периодически беспокоят приступы мучительного непродуктивного кашля, одышка при физической нагрузке. Рецидивирующие пневмонии носят затяжной характер. Увеличивается количество госпитализаций, удлиняются сроки лечения в стационаре. Синусит также приобретает хроническое течение. В полости носа и придаточных пазухах нередко разрастаются полипы. Появляется постоянная заложенность носа.
Из-за хронического кислородного голодания и частых респираторных инфекций страдает общее развитие ребёнка. У таких пациентов снижается аппетит, выявляется недостаточная масса тела, отставание в росте. Наблюдается общая слабость, повышенная утомляемость, ухудшаются способности к обучению. Синдром Картагенера часто является причиной бесплодия у взрослых мужчин.
Осложнения
Острые респираторные заболевания часто встречаются в младшей возрастной группе. По этой причине синдром Зиверта во всём мире выявляется у детей в основном в 3,5–4-летнем возрасте. К этому времени успевают сформироваться бронхоэктазы. Последствия болезни разнообразны. Хроническое воспаление среднего уха приводит к тугоухости или глухоте. Риносинуситы, полипозные разрастания в носу и его придаточных пазухах вызывают резкое снижение обоняния. Бронхоэктазы являются очагом хронической инфекции и провоцируют развитие грозных осложнений. У пациентов с триадой Картагенера часто развивается системный амилоидоз, формируется лёгочно-сердечная и почечная недостаточность. Нередко случаются эпизоды кровохарканья.
Диагностика
Врачи–педиатры первыми сталкиваются с проявлениями триады Картагенера у маленьких пациентов. При осмотре таких детей можно выявить признаки хронической гипоксии. Губы, носогубный треугольник, кончики пальцев приобретают синюшный оттенок. Дистальные фаланги пальцев рук утолщаются подобно барабанным палочкам, ногти приобретают вид часовых стёкол (синдром Мари-Бамбергера). Для окончательного подтверждения диагноза выполняются:
- Физикальное исследование. Перкуторно выявляется смещение кардиальной тупости вправо. При аускультации лёгких выслушиваются жёсткое дыхание и обилие сухих и влажных мелко- и среднепузырчатых хрипов, которые могут исчезать после откашливания. Сердечный толчок и верхушечный тон определяются в 5-ом межрёберном промежутке справа от грудины.
- Лучевая диагностика. На рентгенограмме грудной клетки наблюдаются правостороннее расположение тени сердца, зеркальная транспозиция лёгких. При исследовании назальных синусов часто выявляется недоразвитие фронтальных пазух. Компьютерная томография грудной полости помогает уточнить локализацию и распространённость бронхоэктазий.
- Функциональные методы исследования. Носят вспомогательный характер. На ЭКГ определяется декстрокардия — противоположное направление основных зубцов, признаки лёгочного сердца. При спирометрии (выполняется у детей от 5 лет и старше) выявляется снижение функции внешнего дыхания преимущественно по обструктивному типу.
- Биопсия. Забор материала производится в период ремиссии, не ранее 4-6 недель после купирования обострения. Биоптат слизистой оболочки носа или бронха исследуется с помощью электронной и световой микроскопии. Выявляются аномалии строения цилиарного аппарата, анализируются частота и синхронность колебания ресничек.
Возможно генетическое подтверждение диагноза, однако для синдрома неподвижных ресничек такой метод исследования считается нецелесообразным. Детские пульмонологи дифференцируют синдром Картагенера с муковисцидозом, кистозной гипоплазией лёгких, бронхоэктазами другой этиологии, бронхиальной астмой и первичными иммунодефицитными состояниями.
Лечение синдрома Картагенера
Заболевание генетической природы полностью излечить невозможно. Терапевтические мероприятия выполняются для улучшения качества жизни пациента, сохранения трудоспособности и минимизации последствий. Наследственный синдром является полиорганной патологией. В лечебном процессе также принимают участие оториноларингологи, при необходимости – и другие специалисты. Для первичного подбора базисной терапии лёгочных проявлений показана госпитализация в отделение пульмонологии. Осуществляется длительное консервативное ведение пациента. Используются следующие основные группы лекарственных средств:
- Антибактериальные препараты. При обострении бронхолёгочной патологии и гнойно-воспалительных процессов назальных придаточных пазух необходимы антибиотики. Препарат выбирают с учётом чувствительности микроорганизмов и применяют в зависимости от состояния пациента внутрь или парентерально.
- Кортикостероиды и бронхолитики. Показанием для назначения ингаляционных кортикостероидов, бета-адреномиметиков и холинолитиков является бронхоспастический синдром. При тяжёлой обструкции дыхательных путей применяют препараты системного действия. Топические назальные кортикостероидные гормоны рекомендуются при сочетании полипозного синусита с аллергическим ринитом.
- Муколитики. Назначают профилактическими курсами и при лечении обострений. Предпочтение отдаётся препаратам карбоцистеина, ацетилцистеина, амброксола. Рекомендуется пероральный приём. Исследования использования муколитиков при лечении цилиарных дискинезий в педиатрии доказали неэффективность их ингаляционного введения.
В лечении триады Зиверта-Картагенера широко применяются кинезитерапия, массаж грудной клетки, при необходимости выполняется бронхоальвеолярный лаваж. Иногда для улучшения носового дыхания, аэрации и дренажа назального синуса необходима хирургическая коррекция. В современной оториноларингологии такие операции выполняются преимущественно малоинвазивным эндоскопическим методом. Редко, при выраженных нагноительных процессах осуществляется резекция участка лёгочной ткани. При тяжёлой лёгочно-сердечной недостаточности возможна одномоментная трансплантация комплекса сердце-лёгкие.
Прогноз и профилактика
Прогноз заболевания зависит от распространённости бронхоэктазий, наличия лёгочного сердца и других осложнений. Полного выздоровления не происходит, но своевременная диагностика, чёткое выполнение врачебных рекомендаций позволяют значительно продлить жизнь пациента, улучшить её качество, полностью или частично сохранить трудоспособность.
В качестве первичной профилактики родителям больного ребёнка рекомендуется генетическое обследование перед планированием новой беременности. Пациенту необходимо получать полноценное высококалорийное питание, вести здоровый образ жизни. Для предупреждения обострений показана ежедневная ирригационная терапия – промывание носа и горла солевым раствором. Назначаются курсовые реабилитационные мероприятия. Желательна сезонная профилактическая вакцинация против пневмококка и гриппа.
Источник