Синдром альпорта у детей фото

Синдром Альпорта — редкое генетическое заболевание, характеризующееся прогрессирующей болезнью почек, ушей и глаз. Существует три генетических типа. Наиболее распространенным является синдром X-связанного Альпорта — в этих семьях у пострадавших мужчин, как правило, более тяжелое заболевание, чем у пострадавших женщин. При аутосомно-рецессивном синдроме Альпорта тяжесть заболевания у пострадавших мужчин и женщин аналогична. Существует также аутосомно-доминантная форма, которая поражает мужчин и женщин с равной степенью тяжести. Отличительной чертой заболевания является появление крови в моче в раннем возрасте ребенка, с прогрессирующим снижением функции почек, что в конечном итоге приводит к почечной недостаточности, особенно у мужчин.
Болезнь, которую мы сейчас знаем, как синдром Альпорта, впервые была описана в Британской медицинской литературе в первые годы 20-го века. В 1927 году доктор Сесил Альпорт опубликовал статью, описывающую ассоциацию заболеваний почек и глухоты. Дополнительные случаи были описаны в литературе, и расстройство было названо в честь доктора Альпорта в 1961 году. На фото ниже сам доктор Артур Сесил Альпорт

Альпорт умер в больнице города Лондон в 1959 году, в возрасте 79 лет.
Симптомы синдрома Альпорт
Первый признак болезни почек — кровь в моче. Гематурия обычно не видна невооруженным глазом, но ее можно увидеть, когда мочу осматривают под микроскопом. Это называется микроскопической гематурией. Иногда кровь может быть видна в моче (т. е. моча может быть коричневой, розовой или красной).
Со временем у многих пациентов с синдромом Альпорта обнаруживают повышенный уровень альбумина и других белков в моче, что является показанием того, что болезнь почек прогрессирует. Следующий этап в развитии — постепенное снижение функции почек, часто ассоциируется с высоким кровяным давлением, пока, в конечном счете, почки не перестанут работать. Почки имеют несколько функций включая фильтрацию и выведения продуктов отходов из крови и тела, а также помогает поддерживать баланс некоторых минералов в теле таких как калий, натрий, хлорид, и другие электролиты.
У людей с синдромом Альпорта могут также происходить аномальные изменения в структуре глаза, включая хрусталик, сетчатку и роговицу.
Сетчатка, богатая нервами, светочувствительная мембрана, которая выравнивает заднюю часть глаз, также может быть поражена, как правило, пигментными изменениями, вызванными развитием желтых или белых пятен, поверхностно расположенных на сетчатке. Эти изменения не влияют на зрение. (фото)
Симптомы синдрома могут произойти в кровеносной системе человека, например, аневризмы грудной или брюшной частей аорты. Подробнее про аневризм аорты можно прочесть по статье указанное ниже:
Аневризма — аорты, головного мозга и сосудов мозга, симптомы, лечение операцией
Опубликовано: 08.11.2017
Аневризма расширение артерии, вызванное слабостью в артериальной стенке или ее повреждением. Часто болезнь протекает бессимптомно, но разрыв аневризмы может привести к осложнениям и даже к летальному исходу. Что такое аневризма? Аневризма ослабляет стенки артерии, что создает выпуклость, или разрыв артерии. Выпуклость может иметь две основные формы: Веретенообразные аневризмы дуг всех сторон кровеносного сосуда. Мешотчатые аневризмы
нет комментариев
Причины заболевания
Синдром Альпорта вызван мутациями в конкретных генах. Гены обеспечивают инструкции по созданию белков, которые играют важную роль во многих функциях организма. Когда происходит мутация гена, белковый продукт может быть некачественным, неэффективным, или отсутствовать вовсе. В зависимости от функции конкретного белка, это может повлиять на многие органы и системы организма.
Диагностика синдрома Альпорта
Диагноз синдрома Альпорта ставят на основе выявления характерных симптомов, подробном анамнезе и тщательном клиническом обследовании. Вероятность заболеваемости синдромом Альпорта гораздо выше, если у пациента в семье есть близкие родственники с данным синдромом или с почечной недостаточностью. Различные специализированные тесты могут помочь подтвердить предполагаемый диагноз.
Клиническое обследование
- Биопсия почек. Биопсия почек может выявить характерные изменения в тканях органа, включая нарушения клубочковой фильтрации, которые могут быть обнаружены с помощью электронного микроскопа.
- Анализ мочи. Анализ мочи может выявить количество крови в моче. Если болезнь почек прогрессировала, повышенный уровень белка также может быть обнаружен в образцах мочи.
- Тест на слух. Данная проверка помогает определить звуковой порог вашего слуха. Возраст и продолжительное прослушивание громких звуков могут значительно снизить способность слышать высокие частоты, что является одним из главных симптомов синдрома.
Когда стоит обратиться к врач?
- Если Вы обнаружили кровь при мочеиспускании
- Если Вы заметили, что стали слышать хуже
- Если Вы заметили, что зрение стало хуже
Лечение синдрома Альпорта направлено на конкретные симптомы, которые проявляются у каждого индивидуально. Лечение может потребовать скоординированных усилий группы специалистов. Педиатры, нефрологи, отоларингологи, офтальмологи и другие специалисты здравоохранения, возможно, потребуется систематическое и всесторонне лечение ребенка.
Препараты, известные как ингибиторы ангиотензинпревращающего фермента были использованы для того чтобы обработать пациентов с синдромом Альпорта. Эта терапия не может быть назначена всем пациентам с синдромом Альпорта. Ингибиторы могут быть назначены при повышенном содержании белка. Ингибиторы пациентам с синдромом Альпорта также назначают, что уменьшить протеинурию и заметить прогрессирование болезни почек, задерживая наступление почечной недостаточности.
Организм некоторых людей не реагирует или не переносит ингибиторы группы АПФ. В таком случае пациентам назначают препараты известные как блокаторы рецепторов ангиотензина, которые блокируют действие ангиотензина II, гормона, в норме вырабатываемого Вашими почками.
Диализ — это процедура, в которой машина используется для выполнения некоторых функций почек, помогая контролировать кровяное давление, а также поддерживать надлежащий уровень основных химических веществ, таких как калий.
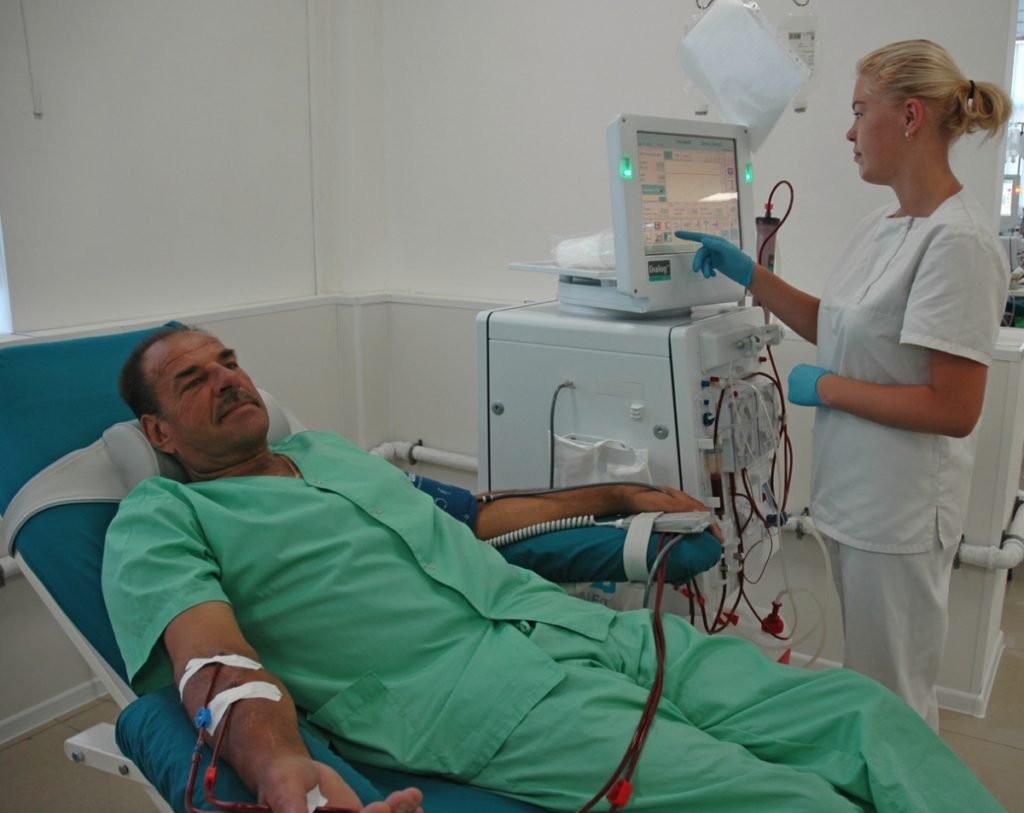
Некоторым пациентами с синдромом Альпорта потребуется пересадка почек в подростковом или юношеском возрасте. Иногда, трансплантация почек является одним из возможных способов лечения.
Источник
Синдром Апера – генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Общие сведения
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.

Синдром Апера
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты. Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост. При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика синдрома Апера
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица. С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения. Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика синдрома Апера
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения. При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов. В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Источник

Добавил:
Вуз:
Предмет:
Файл:
Наследственный нефрит у детей (синдром Альпорта).pptx
Скачиваний:
34
Добавлен:
25.01.2020
Размер:
1.53 Mб
Скачать

АКТУАЛЬНОСТЬ
•Наследственный нефрит является довольно распространенным заболеванием (частота в популяции составляет 1:5000);
•Наследственный нефрит может привести к развитию хронической почечной недостаточности, и как следствие, к летальному исходу;
•При наследственном нефрите поражаются не только почки, но и органы слуха и зрения (может развиться тугоухость вплоть до глухоты).
ОПРЕДЕЛЕНИЕ
Синдром Альпорта (наследственный нефрит) — неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген IV типа базальных мембран, проявляющаяся гематурией и/или протеинурией, прогрессирующим снижением почечных функций, нередко сочетающаяся с патологией слуха и зрения.
ЭПИДЕМИОЛОГИЯ
•Частота СА в популяции составляет 1:5000.
•По эпидемиологическим данным в России частота СА среди детской популяции составляла 17:100000 населения.
•Он служит причиной 1% всех случаев хронической почечной недостаточности (ХПН) в Европе.
КЛАССИФИКАЦИЯ
• По типу наследования:
Х-сцепленный доминантный (классический);аутосомно-рецессивный;аутосомно-доминантный.
• Около 80% в популяции составляют пациенты с Х-сцепленным доминантным вариантом СА, около 15% – с аутосомно-рецессивным СА и около 5% – с аутосомно-доминантным СА.
КЛАССИФИКАЦИЯ
•Тип I – доминантно наследуемый ювенильный тип нефрита с потерей слуха, при котором больные мужчины не могут иметь потомства.
•Тип II – ювенильный тип нефрита с потерей слуха и доминантным,сцепленным с Х-хромосомой наследованием.
•Тип III – «взрослый» тип нефрита с потерей слуха и доминантным,сцепленным с Х-хромосомой наследованием.
•Тип IV – «взрослый» тип нефрита с доминантным, сцепленным с Х-хромосомой наследованием без выраженных изменений слуха.
•Тип V – аутосомно-доминантный нефрит с нарушениями слуха и тромбоцитопатией (синдром Эпштейна).
•Тип VI – ювенильный тип нефрита с потерей слуха и аутосомно- доминантным наследованием.
Ювенильным типом нефрита считается его выявление в возрасте менее 31 года.
*Источник – Редкие заболевания в практике «взрослого» нефролога: наследственный нефрит (синдром Альпорта), болезнь тонкой базальной мембраны, олигомеганефрония. Авторы: И.Г. Каюков, А.М. Есаян, А.В. Смирнов, В.Г. Сиповский, А.Г. Кучер.
ЭТИОЛОГИЯ
•Причина заболевания лежит в мутации одного из генов: COL4A5,
COL4A4, COL4A3.
При классическом варианте болезни мутация происходит в гене COL4A5, расположенном на длинном
плече Х-хромосомы (Хq22.2).
•При одновременной мутации генов COL4A5
и COL4A6 развивается синдром Альпорта с лейомиозом пищевода.
•Мутация, затрагивающая гены
COL4A3 или COL4A4, расположенные на хромосоме 2, характеризуется аутосомно-рецессивным и аутосомно доминантным вариантами наследования.
ПАТОГЕНЕЗ
•В связи с мутацией гена, ответственного за структуру коллагена IV типа, отмечаются изменения в базальных мембранах, прежде всего клубочковых капилляров. Гломерулярная базальная мембрана — сложное образование, которое
состоит из геометрически правильно расположенных молекул коллагена IV типа и полисахаридных компонентов. При мутациях в гене COL4A5 образование спиральной структуры коллагена нарушается.
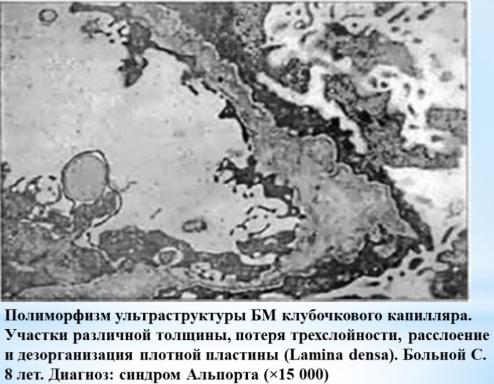
ПАТОГЕНЕЗ
•На первых этапах развития синдрома Альпорта происходит истончение базальной мембраны, особенно ее средней пластинки, одновременно наблюдается расщепление базальной мембраны и появление ее
слоистости. Все это является следствием изменения свойств молекул коллагена.
КЛИНИЧЕСКАЯ КАРТИНА
Для наследственного нефрита (синдрома Альпорта) характерны:
•жалобы на быструю утомляемость, гематурию;
•наличие в родословной случаев синдрома Альпорта или гематурии;
•нарушения слуха и зрения: нарушение слуха может обнаруживаться с помощью аудиометрии, вызванных потенциалов; у больных выявляются различные аномалии зрения, наиболее характерно наличие лентиконуса;
Соседние файлы в предмете Педиатрия
- #
- #
- #
- #
- #
- #
- #
- #
28.11.201822.54 Кб4Новые столы.odt
- #
- #
- #
Источник