Синдром альпорта код мкб

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Гематурический нефрит, Наследственный нефрит 1 типа, Семейный гломерулонефрит.
Названия
Название: Синдром Альпорта.
Синдром Альпорта
Синонимы диагноза
Гематурический нефрит, Наследственный нефрит 1 типа, Семейный гломерулонефрит.
Описание
Наследственное заболевание почек обусловлено изменением синтеза коллагена IV типа, который образует базальные мембраны почечных клубочков, структуру внутреннего уха и хрусталик глаза. Мужчины страдают от тяжелой формы заболевания с выраженными симптомами. Женщины часто являются носителями гена, оставаясь при этом здоровыми, или проявления заболевания у них слабы. Основными симптомами являются микрогематурия, протеинурия, почечная недостаточность, сенсорная потеря слуха, деформация и вывих хрусталика, катаракта. Диагноз ставится на основании клинических и анамнестических данных, результатов общего анализа мочи, биопсии почки, аудиометрии и офтальмологического исследования. Симптоматическое лечение включает лечение ингибиторами АПФ и АРБ.
Синдром Альпорта
Дополнительные факты
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Почти 30 лет спустя, в 1927 году, американский врач А. Альпорт обнаружил частую совместимость гематурии с потерей слуха и уремии у мужчин, тогда как у женщин симптомов не было или была легкой степени. Он предположил наследственную природу заболевания, которое позже назвали синдромом Альпорта. Синонимы — наследственный нефрит типа 1, гематурический нефрит, семейный гломерулонефрит. Распространенность низкая — 1 случай на 5000 человек. Патология представляет 1% пациентов с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почки. Заболевание диагностируется у людей всех рас, но соотношение разных форм не одинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием — его развитие обусловлено дефектом генов, которые кодируют структуру нескольких коллагеновых цепей типа IV. Генетические изменения представлены делециями, сплайсингом, бессмысленными и бессмысленными мутациями. Его расположение определяет тип наследования заболевания:
• Доминанта с X-связью. Это связано с мутацией в локусе COL4A5, расположенном на хромосоме X. Ген кодирует цепь коллагена a5 типа a5. Этот генетический дефект вызывает 80-85% случаев наследственного нефрита. Болезнь полностью проявляется у мальчиков и мужчин, у женщин оставшийся нормальный ген на Х-хромосоме компенсирует выработку функционального коллагена.
• Аутосомно-рецессивный. Он развивается на основе мутаций в генах C0L4A3 и COL4A4. Они расположены на второй хромосоме и отвечают за структуру коллагеновых цепей а3 и а4. Пациенты с этим вариантом синдрома составляют около 15% пациентов. Тяжесть симптомов не зависит от пола.
• Аутосомно-доминантный. Нефрит возникает в результате мутаций в генах COL4A3-COLA4, расположенных на хромосоме 2. Как и в случае аутосомно-рецессивной формы заболевания, синтез цепей коллагена а4 и а3 четвертого типа нарушается. Распространенность — 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложную структуру, она образует строгую геометрическую последовательность молекул коллагена 4-го типа и полисахаридных компонентов. Синдром Альпорта имеет мутации, которые определяют дефектную структуру спиральных молекул коллагена. На первых этапах заболевания базальная мембрана истончается, начинает расщепляться и отслаиваться. В то же время есть утолщенные участки с неравномерным освещением. Мелкозернистое вещество накапливается внутри. Прогрессирование заболевания сопровождается полным разрушением базальной клубочковой оболочки, клубочковых капилляров, почечных канальцев, структур внутреннего уха и глаз. Таким образом, патогенетический синдром Альпорта представлен четырьмя связями: мутация гена, дефект в структуре коллагена, разрушение базальных мембран и патология почек (иногда нарушения слуха и зрения).
Симптомы
Наиболее распространенным проявлением синдрома Альпорта является гематурия. Этот симптом наблюдается под микроскопом у 95% женщин и 100% мужчин. При рутинном осмотре мальчиков гематурия обнаруживается в первые годы жизни. Другим распространенным признаком заболевания является протеинурия. Выведение белка с мочой у мужчин с синдромом Х-хромосомы начинается в раннем детстве, остальные — позже. Выделение белка несколько увеличивается у девочек и женщин, а случаи выраженной протеинурии встречаются крайне редко. У всех пациентов наблюдается устойчивое прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоего пола с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается с увеличением ХПН. У юношей, мужчин, снижение функции почек достигает конечной стадии к 16-35 годам, при медленном течении заболевания — к 45-65 годам. Иногда обнаруживаются диффузные опухоли гладких мышц пищевода и бронхов, которые в позднем детстве проявляются дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой и частым бронхитом.
Нередко у пациентов формируется нейросенсорная тугоухость. Нарушение слуха начинается в детстве, но становится заметным в подростковом или юношеском возрасте. У детей потеря слуха распространяется только на высокочастотные звуки; обнаруживается в специально созданных условиях — во время аудиометрии. По мере старения и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, включая человеческую речь. При синдроме Х-сцепления потеря слуха через 25 лет наблюдается у 50% больных мужчин, через 40 лет — у 90%. Тяжесть потери слуха варьируется от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
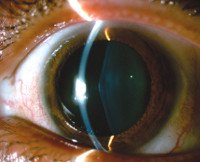
Нарушения зрения включают передний лентиконус, выпячивание хрусталика центра глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением и болью в глазах. У некоторых пациентов наблюдается стигма дисембриогенеза: анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может быть высокое положение неба, укорачивание и изгиб мизинцев, суставы пальцев, хорошо разнесенные глаза.
Одышка. Рвота. Судороги. Тремор.
Возможные осложнения
Отсутствие лечения для пациентов с синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, образованию катаракты. У некоторых пациентов развивается полинейропатия — повреждение нервов, сопровождающееся мышечной слабостью, болью, судорогами, тремором, парестезией и снижением чувствительности. Еще одним осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным заболеванием при наследственном нефрите является конечная стадия почечной недостаточности. Больше всего это затрагивает мужчин с той или иной формой наследования, связанной с сексуальной Х-хромосомой. В возрасте 60 лет 100% пациентов этой группы нуждаются в гемодиализе, перитонеальном диализе и трансплантации почки.
Диагностика
Нефрологи, урологи, терапевты и генетики участвуют в диагностическом процессе. Опрос показывает возраст появления симптомов, наличие у родственников первой линии гематурии, протеинурии или смерти от хронической почечной недостаточности. Синдром Альпорта характеризуется ранним началом и напряженной семейной историей. Дифференциальный диагноз направлен на устранение гематурической формы гломерулонефрита, вторичной нефропатии. Для подтверждения диагноза выполняются следующие процедуры:
• Физическое обследование. Определяются бледность кожи и слизистых оболочек, снижение мышечного тонуса, внешние и соматические признаки дисембриогенеза — высокое небо, нарушения в строении конечностей, увеличение расстояния между глазами, сосками. Артериальная гипертензия диагностируется на ранних стадиях заболевания, а артериальная гипертония — на поздних стадиях.
• Общий анализ мочи. Эритроциты и высокий уровень белка обнаружены — признаки гематурии и протеинурии. Индекс мочевого белка напрямую коррелирует с тяжестью синдрома, развитием патологии, вероятностью нефротического синдрома и хронической почечной недостаточностью, оцениваемой на основании его изменения. Могут быть признаки бактериальной лейкоцитурии.
• Биопсия почки. Микроскопия визуализирует истончающуюся базальную мембрану, расщепляя и разделяя ее слои. На поздней стадии отмечались концентрированные дистрофические участки с «клетками» просветления, зоны полного разрушения пласта.
• Молекулярно-генетическое тестирование. Генетическая диагностика не является обязательной, но она позволяет более точный прогноз, выбирая оптимальную схему лечения. Мы изучаем структуру генов, в которых мутации вызывают развитие команды. Мутации в гене COL4A5 обнаружены у большинства пациентов.
• Аудиометрия, офтальмологическое обследование. Кроме того, пациентам могут быть назначены диагностические консультации у аудиолога и офтальмолога. При аудиометрии выявляется потеря слуха: в детском и подростковом возрасте — двусторонняя высокочастотная потеря слуха, во взрослом возрасте — низкочастотная и среднечастотная потеря слуха. Офтальмолог определяет искажение хрусталика, повреждение сетчатки, катаракту и снижение зрения.
Лечение
Специальной терапии нет. С раннего возраста проводится активное симптоматическое лечение, которое уменьшает протеинурию. Это позволяет предотвратить повреждение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов ангиотензина II можно остановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией хронической почечной недостаточности назначают гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почки.
Список литературы
1. Наследственный нефрит (синдром Альпорта) / Сунгатуллина И. Л. // Казанский медицинский журнал – 2002 – Т. 83, №1.
2. Проект клинических рекомендаций по диагностике и лечению синдрома Альпорта у детей / Длин В. В. , Игнатова М. С. , Конькова Н. Е. – 2014.
3. Наследственные заболевания почек, протекающие с гематурией / Длин В. В. , Игнатова М. С. // Российский вестник перинатологии и педиатрии – 2014 — №3.
Источник

Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Синдром Альпорта
Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН. У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам. Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
Источник