Синдром айкарди что это такое

Синдром Айкарди является редким цереброретинальным генетическим расстройством. При данном заболевании мозг, частично или полностью, лишен одной из основных составляющих – мозолистого тела. Это является следствием дефекта Х-хромосомы. В сетчатке происходят изменения, ведущие к инфантильным спазмам.
Мозолистое тело – центральный отдел нервной системы, представленный нервными окончаниями, соединяющими полушария головного мозга.
Сидром Айкарди не является отдельной формой эпилепсии, а является самостоятельным заболеванием, которое в 1965 году было описано невропатологом из Франции Жаном Экарди.
Синдром Айкарди-Гутьереса является угрозой для мальчиков с хромосомным набором ХХY или с синдромом Клайнфельтера.
По данным статистики, на сегодняшний момент наблюдалось около 500 случаев этого заболевания. Наибольшее распространение оно получило в Японии.
Однако, национальности и расы не играют ключевой роли. Все зависит от уровня медицины в регионе. Недостаток знаний ведет к не выявленным случаям. Из 2 – 4% детей, у которых проявляются инфантильные спазмы, обнаруживается болезнь Экарди.
Этиология и патогенез синдрома
До сих пор точно не установлена причина данного заболевания. Соответственно, невозможно определить факторы риска.
Специалисты отвергают наследственность. Если в семье есть ребенок с данным синдромом, то шанс рождения второго с такой же болезнью составляет меньше 1%.
Выдвигается следующая теория. Клетки эмбрионов содержат по одной активной Х-хромосоме. Следовательно, их инактивация при нормальном развитии происходит случайно.
При синдроме Айкарди, случайной инактивации не наблюдается и большинство клеток формируется при участии только этой хромосомы.
Какой ген отвечает за эти изменения к настоящему времени неизвестно. Предположительно, это мутированный de novo на инактивированной Х-хромосоме.
Как проявляет себя синдром?
Дети 2 – 5 месяцев, у которых обнаруживается синдром Айкарди, внешне не отличимы от здоровых малышей. Позже начинают проявляться судороги или инфантильные спазмы. Они представляют вид эпилептического синдрома.
Ребенок снижает активную деятельность и впадает в ступор. Взгляд прикован к одной точке. Руки начинают подниматься вверх и сгибаться.
Тело выгибается и ножки начинают резко выпрямляться. На это уходят секунды и продолжения можно ожидать в любое мгновение. Во время припадков, малыш становится раздражителен и постоянно плачет. Позже, такие судорги часто ведут к эпилепсии.
Параллельно присутствуют и другие симптомы синдрома Айкарди:
- поражается сетчатка, в глазах появляются желтоватые пятна;

На фото глазное яблоко при синдроме Айкарди
- глаза ребенка ненормально-малого размера;
- врожденная колобома – выемка, щель или прорезь в радужной оболочке глаза;
- задержка развития;
- проблемы при кормлении ребенка;
- диарея;
- попадание пищи и желудочного сока в пищевод – гастроэзофагеальный рефлюкс (у взрослых-изжога). Часто проходит самостоятельно и не является серьезным заболеванием. Однако следует знать, что это один из признаков заболевания;
- мышечная апатичность.
По наблюдениям за больными можно выделить и дополнительные признаки:
- латеральное расположение бровей, их редкость;
- выступающие резцы и вздернутый нос;
- уменьшен угол носовой перегородки.
Иногда наблюдаются наросты и уплотнения на коже: невусы, дивертикулы кожи и опухоли, причиной которых является патология кровеносных сосудов – гемангиомы. Редко встречается аномалия рук.
Встречаются заболевшие с плагиоцефалией (сплюснутая область на голове), с ассиметрией лица, а также с расщелинами неба или верхней губы. Размеры головы и рук заметно меньше, чем у здорового человека. Нос плоский, а уши чрезмерно большие. Нос и губы разделяет слишком малое пространство.
У большего количества больных происходило развитие тяжелых эпилептических припадков, которые не проходили до конца жизни. Известно наличие полу-позвонков и отсутствия ребер, приводящее к заметному искривлению позвоночника.
Увеличен риск развития опухолей:
- папиллом сосудистого сплетения;
- липом;
- ангисарком;
- гепатобластом;
- гемангиом;
- ангиосарком;
- гепатобластом;
- интерстициального полипоза;
- эмбриональной кальциомы;
- полипоза кишечника.
 Аномалии в глазах могут привести к пигментному рениту, микрофтальми. Возможна катаракта и атрофия зрительного нерва. Это может привести к нарушению зрения и даже полной слепоте.
Аномалии в глазах могут привести к пигментному рениту, микрофтальми. Возможна катаракта и атрофия зрительного нерва. Это может привести к нарушению зрения и даже полной слепоте.
Нередко нарушается эндокринная система – позднее наступление половой зрелости или явная задержка.
Физическое развитие начинает замедляться. В возрасте 7-10 лет больной выглядит как пятилетний. Вес увеличивается также с задержкой.
Согласно исследованиям ученых, в мозге здорового ребенка больше складок, чем у болеющего синдромом Айкарди. Случается, в пораженном мозге образуются кисты, которые заполнены жидкостью.
Большинство заболевших имеют явно выраженную задержку в умственном развитии, однако, иногда они просто плохо способны к обучению.
Способность к членораздельной речи часто развита очень слабо. Самостоятельное передвижение наблюдается редко. Некоторые полностью зависимы от помощи других людей.
Постановка диагноза
На сегодняшний момент нет специальной технологии диагностики, с помощью которой можно точно поставить диагноз. Как правило, применяются следующие методы:
- Неврологический осмотр, в процессе которого врач оценивает состояние нервной системы ребенка и определяет ее расстройства.
- Офтальмоскопия включает в себя изучение глазного дна, его сосудов и сетчатки.
- Электроэнцефалограмма (ЭЭГ). С ее помощью исследуют работу головного мозга, происходит регистрация электрических импульсов, которые исходят из его отдельных частей. Является основным методом для диагностики эпилепсии и других заболеваний. При синдроме Айкарди помогает определить тяжесть и тип приступов.
- Магнитно-резонансная томография (МРТ). Основу этого диагностического исследования составляет постоянное магнитное поле, быстро меняющиеся локальные магнитные поля и радиочастотная энергия. Специально предназначенная для этого аппаратура создает четкое изображение внутреннего органа. С помощью обнаруживается патология мозолистого тела, ассиметрия полушарий, внутримозговая киста и другие аномалии.
- Компьютерная томография (КТ). С ее помощью специалисты выявляют участки мозга, которые подверглись повреждениям в результате болезни.
- Рентген скелета.
- Генные исследования.

Другие диагностические методы могут проводиться в зависимости от симптомов и общего состояния больного.
Симптоматическое лечение — единственный вариант
На сегодняшний момент комплексного лечения синдрома Айкарди нет. Применяется терапия каждого симптома.
Принятая тактика – купировать инфантильные спазмы, являющиеся резидентными к антиэпилептическим препаратам. Используются максимально высокие дозы разнообразных медикаментов.
После выявления заболевания, на первых порах используется Вигабатрин, он же Сабрил. Его применяют от 50 до 100 мг на килограмм веса в сутки. Параллельно с ним вводятся вальпроаты. Их суточная норма – 50–100 мг на кг.
Если приступы учащаются, то к АЭП добавляются бензодиазепины, от 0,25 до 2 мг в сутки. Нередко применяется Фенобарбитал или Суксилеп.
В качестве альтернативы выступают кортикостероидные гормоны. Это:
- АКТГГ;
- Синактен-депо;
- Дексаметаз;
- Преднизолон;
- Октагам.
Поначалу Преднизалон назначается из рассчета 1–2,5 мг на кг, затем эта доза снижается до щадящей. Как правило, гормоны применяются параллельно базовым АЭП.
К сожалению, эти методы малоэффективны и редко достигают полного подавления синдрома.
В виде палиативного хирургического лечения может быть стимулирован блуждающий нерв. Дефекты костей, которые способствуют сколиозу, устраняются физиотерапией, лечебной физкультурой или исправляются при помощи хирургической коррекции.
Разработаны специальные программы, противодействующие умственной отсталости. Для длительного лечения патологических состояний необходима помощь детского невропатолога.

Прогноз и летальность
Прогноз при синдроме Айкарди достаточно неблагоприятный и зависит от тяжести спазмов и сопровождающих их заболеваний:
- смертность в детском возрасте – 25%;
- выживают и могут самостоятельно ходить – 25%;
- могут самостоятельно себя обслуживать – 50%.
Больные живут приблизительно от 9 до 19 лет. Однако известны случаи, когда заболевшие женщины прожили до 32 и 49 лет (умеренная форма синдрома).
Несмотря на редкость заболевания, ему посвящен отдельный сайт (www.aicardisyndrome.org). На нем можно пообщаться с другими людьми, страдающими этим заболеванием, и их семьями. Задать интересующие вопросы, поделиться опытом и другое. Здесь можно получить информацию о благотворительных мероприятиях, организатором которых является Фонд Экарди.
К сожалению, в связи с мало изученностью данного заболевания, профилактических мер против синдрома Айкарди пока не существует.
Источник
Синдром айкарди является редким генетическим заболеванием, при котором наблюдается агенезия в мозолистом теле. В период протекания заболевания диагностируются эпилептические приступы, которые по своему типу напоминают инфантильные спазмы.
Причины и симптомы болезни
Причины миоклонической энцефалопатии на сегодняшний день не установлены. Заболевание развивается у новорожденных малышей. В возрасте ребенка 2-5 месяцев признаки не наблюдаются. По внешнему виду пациенты не отличаются от здоровых детей.
После достижения 6-месячного возраста синдром айкарди сопровождается судорогами и инфантильными спазмами. У ребенка диагностируется снижение активной деятельности и развитие ступора. Малыш концентрирует свой взгляд на одной точке.

Миоклоническая энцефалопатия сопровождается резким выгибанием тела малыша и выпрямлением ножек. Припадки сопровождаются раздражительностью и постоянным плачем малыша. При судорогах у маленьких пациентов диагностируют развитие эпилепсии.
Синдром айкарди сопровождается дополнительной симптоматикой. Заболеванием поражается сетчатка глаза, что приводит к возникновению пятен желтоватой окраски. Органы зрения ребенка имеют ненормально малый размер.
Патология сопровождается врожденной коломбой, которая характеризуется наличием щели, выемки или прорезью в радужной оболочке. В период кормления малыша появляются сложности. Миоклоническая энцефалопатия сопровождается диареей.
В пищевод попадает пища или желудочный сок, что приводит к развитию гастроэзофагеального рефлюкса. Такой симптом проходит самостоятельно и не требует медицинского вмешательства. При синдроме айкарди диагностируют мышечную апатичность.

Если наблюдать за пациентом, то это позволит определить развитие дополнительной симптоматики. Брови у малыша расположены латерально. Нос ребенка является вздернутым, а резцы – чрезмерно выступающими. Угол носовой перегородки при синдроме айкарди незначителен.
У пациентов миоклоническая энцефалопатия сопровождается наростами и уплотнениями на кожных покровах – дивертикулами, невусами, опухолевыми процессами, которые развиваются на фоне патологии кровеносных сосудов – гемангиом. В редких случаях при синдроме айкарди диагностируют аномалии верхних конечностей.
У детей заболевание диагностируется по ассиметрии лица. В области верней губы и неба имеются расщелины. Голова и руки имеют меньшие размеры, чем у здорового ребенка. Между носом и губами имеется небольшое пространство.
На фоне аномалий в глазах у пациентов развивается при синдроме айкарди микрофтальм и пигментный ринит. Возможно развитие атрофии зрительного нерва и катаракты. У больных нарушается зрение, что становится причиной полной слепоты.

Миоклоническая энцефалопатия сопровождается нарушениями в работе эндокринной системы. У детей поздно наступает половая зрелость или наблюдают ее явную задержку. Синдром айкарди сопровождается замедлением физического развития. Увеличение веса наблюдается с задержкой.
Миоклоническая энцефалопатия сопровождается большим количеством симптомов, при возникновении которых рекомендовано провести обследование для назначения действенного лечения.
Диагностические мероприятия
Специальная технология диагностики синдрома айкарди отсутствует. Проведение диагностики требует прохождения нескольких этапов, которые заключаются в:
- Неврологическом осмотре. Врачом оценивается работоспособность нервной системы и определение расстройств.
- Офтальмоскопии. Этот метод исследования позволяет изучить глазное дно и сетчатку.

- Магниторезонанской томографии. Диагностическая процедура требует применения постоянного магнитного поля и радиочастотной энергии. Благодаря использованию специально предназначенной аппаратуры обеспечивается возможность получения четкого изображения внутреннего органа.
В период проведения манипуляции обеспечивается возможность определения патологии в мозолистом теле, внутримозговой кисты, ассиметрии полушарий.
- Электроэнцефалограммы. При синдроме айкарди проводится исследование работы головного мозга, регистрации электроимпульсов, которые издаются определенными частями головного мозга. Метод используется при миоклинической энцефалопатии для определения типа и тяжести приступов.

- Компьютерной томографии. Предоставляет возможность определения участков мозга, которые повреждены синдромом айкарди.
Если имеются подозрения на миоклинический тип заболевания, то рекомендовано проведение генных исследований и рентгена скелета.
Диагностика синдрома айкарди требует использования нескольких методов, что позволит точно определить степень поражения органа.
Лечение и прогноз
Методы терапии синдрома айкарди на данный момент не разработаны. Пациентам рекомендовано применение симптоматического лечения. Основным методом терапии миоклонической энцефалопатии является купирование симптоматики.
Изначально при синдроме айкарди назначают вигабатрин (Сабрил). Пациенту рекомендовано принимать препарат из расчета 50-100 миллиграмм на килограмм веса. Одновременно миоклоническая энцефалопатия излечивается с применением вальпроатов.

Пациентам делают назначение Депакин-сиропа в количестве 50-100 миллиграмм на куилограмм веса в день. Если синдром айкарди сопровождается частыми приступами, то рекомендовано комбинировать АЭП и бензодиазепины.
Рекомендован прием Клоазепама в количестве 0,25-2 миллиграмма в сутки. При синдроме айкарди пациентам прописывают Фенобарбитал – 5-15 миллиграмм на килограмм массы пациента в сутки.
Если протекает миоклоническая энцефалопатия, то лечение проводится Суксилепом. Рекомендовано применение 15-30 миллиграмм препарата на килограмм массы тела ребенка в день. Пациентам рекомендуют использовать кортикостероидные гормоны: Преднизолон, Дексаметазон, Синактен-депо и т.д.

Преднизолон назначается в количестве 1-2,5 миллиграмм на килограмм массы пациента. После достижения положительного терапевтического эффекта рекомендовано использование поддерживающей дозы препарата.
При недостаточной эффективности медикаментозного лечения назначается хирургическое вмешательство. Пациентам рекомендуется проводить стимуляцию блуждающего нерва. Во избежание развития сколиоза рекомендовано проведение при синдроме айкарди лечебной физкультуры, физиотерапии. При возникновении необходимости проводят хирургическую коррекцию.
Миоклоническая энцефалопатия характеризуется неблагоприятным прогнозом. Он напрямую зависит от степени тяжести патологии. В 25 процентов случаев наблюдается летальный исход у пациентов с синдромом айкарди в детском возрасте.

При заболевании самостоятельно ходит 25 процентов человек, 50 процентов больных могут самостоятельно себя обслуживать. Длительность жизни пациентов составляет 9-19 лет. При умеренной форме синдрома пациент может прожить до 49 лет.
Миоклоническая энцефалопатия является редким генетическим заболеванием, которое не поддается лечению. Патология диагностируется у новорожденных детей. Первые признаки заболевания проявляются у пациентов 5-месячного возраста.
При возникновении симптомов патологии рекомендовано обратиться за помощью к доктору. После проведения соответствующих диагностических процедур специалист разработает действенное лечение, которое будет направлено на снижение выраженности симптоматики. Специфической профилактики заболевания не существует, так как его причины не установлены.
Источник
Что такое синдром Айкарди?
Синдром Айкарди — крайне редкое генетическое заболевание. Почти все люди с синдромом Айкарди — новорожденные девочки. У людей с синдромом Айкарди наблюдается агенезия мозолистого тела (отсутствие соединения между двумя полушариями мозга), хориоретинальные лакуны (дефекты в светочувствительной ткани в задней части глаза) и эпилептический припадок.
Синдром является спорадическим (случайным), что означает, что оно не передается от родителя к ребенку.
Причины
Синдром Айкарди, вероятно, возникает в результате новой мутации в гене, расположенном на Х-хромосоме. Точный ген или генетический механизм, вызывающий синдром Айкарди, пока неизвестен. Недавно обнаруженный отчет, описывающий изменения в генах TEAD1 и OCEL1 у двух девочек с синдромом, не был подтвержден в большой группе других девочек с синдромом Айкарди. Таким образом, эти гены, по-видимому, не являются причиной синдрома Айкарди. Предполагается, что это состояние является смертельным для новорожденных детей мужского пола.
Родители женщины с синдромом Айкарди обычно не страдают. О передаче синдрома Айкарди от пострадавшей матери ее ребенку не сообщалось. Другие члены семьи также обычно не подвергаются повышенному риску.
Симптомы и признаки
Синдром Айкарди обычно начинается с непроизвольных мышечных спазмов в возрасте от четырех месяцев до четырех лет.
Другие симптомы могут включать эпилепсию, умственную отсталость, глубокую мышечную слабость (гипотонию), аномально маленькую голову (микроцефалию), аномально маленькие глаза (микрофтальмию), неполное развитие сетчатки и нерва в задней части глаза (колобомы) и/или аномалии ребер и/или позвоночника.
У детей всех возрастов с синдромом Айкарди наблюдается значительная задержка моторного развития. Синдром Айкарди может быть опасным для жизни в детстве из-за осложнений инфекций верхних дыхательных путей.
Большая часть девочек с синдромом Айкарди имеет резкую задержку психомоторного развития. В неврологическом статусе часто отмечается — значительное уменьшение размеров черепа (микроцефалия), мышечная гипотония, возможна односторонняя мышечная гипертония и спастичность, оживленные глубокие сухожильные рефлексы или нарушение работоспособности конечностей (геми- или тетрапарез).
Агенезия мозолистого тела при синдроме Айкарди обычно тотальная, часто сопровождается с гетеротопией коркового вещества мозга, атрофией коры, структурной асимметрией полушарий мозга, нормотензивной гидроцефалией, полимикрогирией или пахигирией, хориоидальными кистами и папилломами, вентрикуломегалией, внутримозговыми кистами, синдромом Денди-Уокера.
Затронутые группы населения
Известно приблизительно о 500 случаях синдрома Айкарди во всем мире, особенно большое количество в Японии. Синдром встречается у детей с различной расовой принадлежностью.
По недавно проведенным в Швеции исследованиям распространенность синдрома Айкарди составляет от 2 до 15 случаев на 100000 девочек. В России аналогичные исследования к сожалению не проводились. Однако, учитывая фенотипическое разнообразие и диагностические трудности, многие случаи заболевания остаются недиагностированными. Это позволяет пересмотреть данные об истинной распространенности синдрома Айкарди в сторону увеличения, возможно, синдром Айкарди является более частой причиной задержки умственного развития и инфантильных спазмов у девочек, чем считается в настоящее время.
На сегодняшний день считается, что заболеваемость синдромом Айкарди среди всех детей с инфантильными спазмами составляет всего около 2-4%.
Диагностика
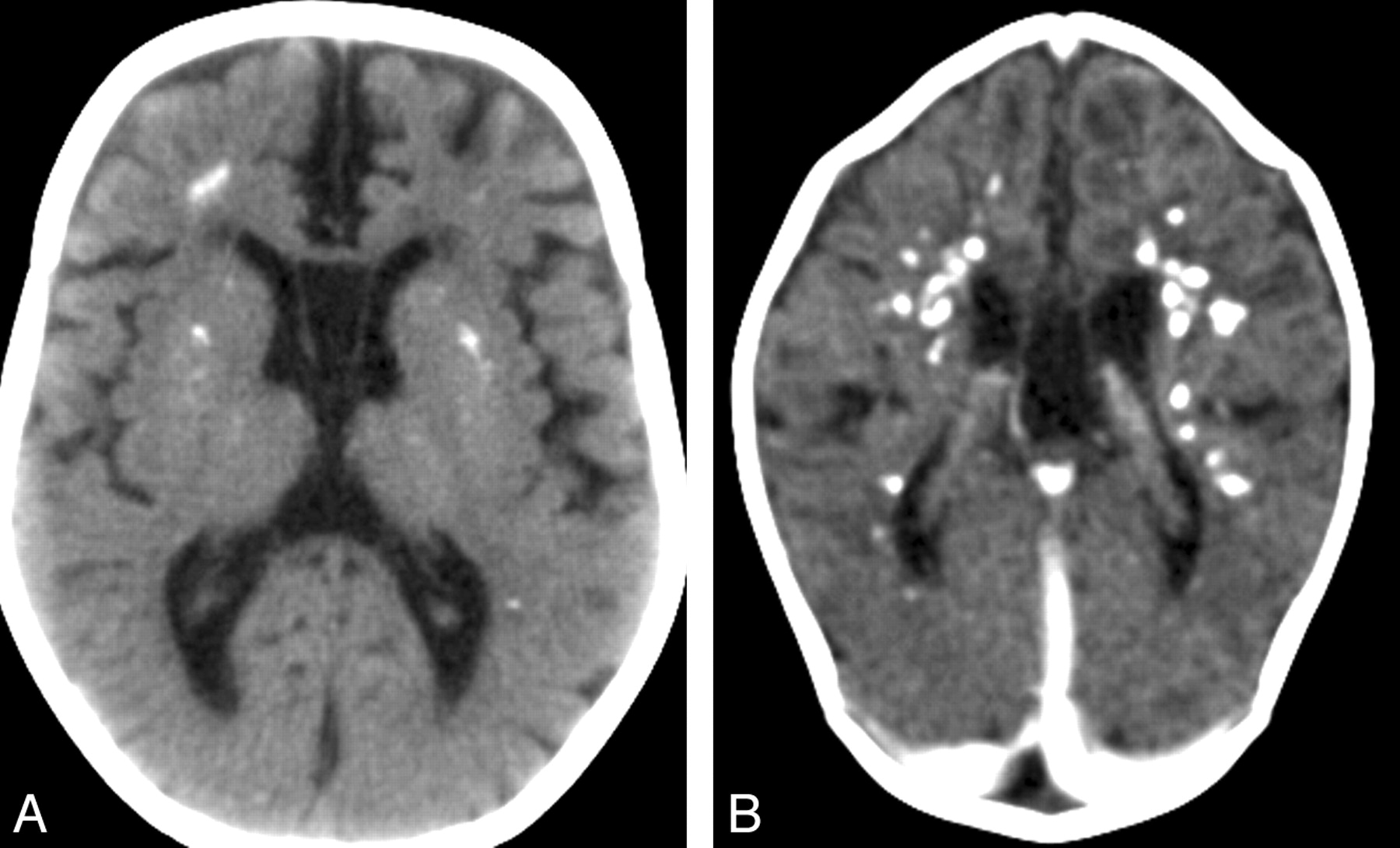
До настоящего времени не существует специального лабораторного диагностического теста или исследования, которое бы позволило поставить диагноз синдрома Айкарди. Для этого необходимо:
- неврологический осмотр;
- офтальмоскопия;
- электроэнцефалография (ЭЭГ);
- магнитно-резонансная томография с контрастом и/или без;
- рентгенограмма скелета.
На магнитно-резонансной томографии можно обнаружить агенезию мозолистого тела, асимметрию полушарий коры, гетеротопию коркового вещества, внутримозговые кисты, папиллому сосудистых сплетений и тд.. Аксоны коры, которые в норме должны перекрещиваться, при его агенезии не формируются и соответственно не идентифицируются при нейровизуализации.
Агенезия мозолистого тела позволяет боковым желудочкам распространиться вверх, во фронтальное и париетальное белое вещество. Это состояние именуется верхней транслокацией боковых желудочков в лобно-теменные регионы мозга. Аналогичное смещение вверх претерпевает и третий желудочек, что является одним из нейрорадиологических маркеров агенезии мозолистого тела. Увеличенный третий желудочек, выдвигаясь вперед и вверх, раздвигает передние рога боковых желудочков, при сопутствующей гидроцефалии объем желудочков увеличивается, задние рога расширяются и изгибаются по направлению к средней линии (форма «ухвата»). Вероятно отсутствие поддерживающей функции мозолистого тела является основой для типичной черты агенезии мозолистого тела — расширения полушарий, третьего желудочка и Монроева отверстия.
Типичные изменения на электроэнцефалографии в виде гипсаритмии, характерные для инфантильных спазмов встречаются далеко не у всех больных. Наиболее характерные изменения приступной ЭЭГ заключаются во вспышках нерегулярных быстрых и медленных волн продолжительностью от 3 до 6 секунд, которые перемежаются некоторым уплощением основного ритма в течение 5-20 секунд, причем изменения не синхронизированы по полушариям. На электроэнцефалографии — феномен «расщепленного мозга». Так как практически у половины (42%) пациентов инфантильные спазмы сочетаются с другими видами эпилептических приступов, то данные ЭЭГ могут быть противоречивы.
При офтальмоскопии обнаруживаются белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки.
Лечение синдрома Айкарди
Лечение синдрома Айкарди пока не разработано. В основном применяется симптоматическое лечение. Основная стратегия терапии — купирование инфантильных спазмов, которые зачастую резистентны к антиэпилептическим препаратам, лечение их сложное и эффективность его невелика. Применяют различные медикаменты в максимально высоких дозах.
Стартовая терапия начинается с вигабатрина (сабрил) — 50-100мг/кг/сут и вальпроатов (депакин-сироп) — 50-100мг/кг/сут. При частых приступах назначают комбинации антиэпилептических препаратов с бензодиазепинами (клоназепам) -0,25-2мг/сут, или фенобарбиталом(5-15мг/кг/сут), а так же может быть введен суксилеп 15-30мг/кг/сут.
Альтернативным методом является применение кортикостероидных гормонов (АКТГГ, синактен-депо в/м, дексаметазом, преднизолон) и октагам. Средняя дозировка преднизолона 1-2,5мг/кг/сут с последующим переходом на минимальную поддерживающую дозу. Гормоны назначают в сочетании с базовыми антиэпилептическими препаратами.
В качестве паллиативного хирургического лечения возможно использование стимуляции блуждающего нерва. Так как костно-мышечные дефекты могут приводить к сколиозу, для его предотвращения используется физиотерапия, лечебная физкультура, возможна хирургическая коррекция.
Прогноз
Прогноз для девочек с синдромом Айкарди варьируется в зависимости от тяжести их симптомов.
Прогноз при синдроме Айкарди, как правило, серьезен в связи с выраженной умственной отсталостью и резистентным характером судорог. Часть детей (до 25%) погибает в первые годы жизни. Из выживших детей только 25% самостоятельно ходят и только 50% имеют навыки самообслуживания.
Продолжительность жизни очень вариабельна, в зависимости от степени выраженности симптомов. Средняя продолжительность жизни по разным данным составляет от 8,3 до 18,5 лет. Но имеются сведения о женщине 32 лет с синдромом Айкарди, а так же 49 лет с умеренной формой синдрома
Источник