Широкие дистальные фаланги и гримаса улыбки характерны для синдрома

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 30 сентября 2013;
проверки требуют 11 правок.
Синдром Рубинштейна — Тейби (синоним: синдром широкого I пальца кистей и стоп, специфического лица и умственной отсталости) впервые описан в 1963 г. J. Rubinstein и Н. Taybi[2].
Популяционная частота — 1:25 000 — 30000, соотношение полов — 1:1.
Тип наследования аутосомно-доминантный (ген CREBBP в локусе 16p13.3), но в большинстве случаев мутация возникает спонтанно, то есть не наследуется от родителей[3][4].
Дифференциальный диагноз: брахидактилия, тип D, акроцефалосиндактилия, тип VI.
Минимальные диагностические признаки: прогрессирующая умственная отсталость, широкие терминальные фаланги первых пальцев кистей и стоп, характерное лицо, отставание роста и костного возраста, микроцефалия, крипторхизм.
Клиническая характеристика[править | править код]
Основными признаками синдрома являются отставание психофизического развития, дисплазия лица, аномалии развития пальцев. У 94 % больных рост ниже среднего. Костный возраст значительно отстаёт от паспортного (94 %). Отмечается олигофрения (100 %) (в большинстве случаев — тяжёлая умственная отсталость, задержка моторного и речевого развития). Характерна чрезмерная отвлекаемость, плохая концентрация внимания, трудность овладения речью[5].
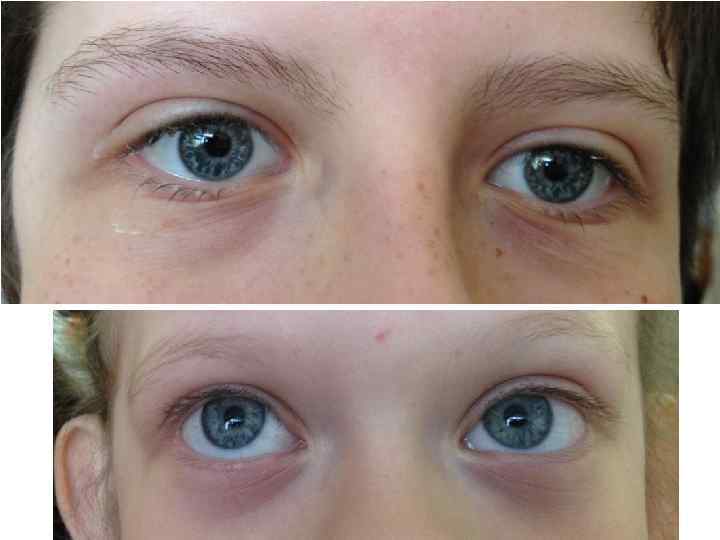
Комплекс черепно-лицевых дисморфий включает брахицефалию, микроцефалию, большой и поздно закрывающийся родничок, выступающий лоб с низким ростом волос, приподнятые дугообразные брови, антимонголоидный разрез глаз (93 %), широкую переносицу, эпикант (62 %), длинные ресницы, птоз, широкую спинку носа (71 %), загнутый книзу кончик носа (клювовидный нос) (90 %), гипоплазию крыльев носа (72 %), умеренную ретрогнатию, гримасу, напоминающую улыбку, высокое арковидное небо (93 %).
Со стороны глаз отмечаются косоглазие (79 %) и аномалии рефракции (58 %). Ушные раковины деформированы, уменьшены или увеличены (74 %).
Аномалии пальцев заключаются в расширении, укорочении и уплощении ногтевых фаланг первых пальцев кистей и стоп (100 %), иногда терминальных фаланг других пальцев кисти (60 %), вальгусной деформации межфаланговых суставов, удвоении ногтевой фаланги первых пальцев стоп (30 %), реже — проксимальной фаланги, в ряде случаев — полидактилии стоп, частичной синдактилии кистей и стоп.
Отмечаются также лордоз, кифоз, сколиоз, аномалии грудины и рёбер, уплощение крыльев тазовых костей.
В половине случаев встречаются гирсутизм, ярко-красный невус на коже лба, затылка, боковой поверхности шеи. Часты изменения дерматоглифических показателей.
Пороки развития внутренних органов включают незаращение артериального протока и дефекты перегородок сердца, одностороннюю аплазию почек, удвоение почек, гидронефроз, расширение мочеточников или их стеноз, дивертикул мочевого пузыря, крипторхизм (в 79 % случаев), нарушение лобуляции легких, агенезию или аплазию мозолистого тела (обнаруживаемую при МРТ-исследовании).
Примечания[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ RUBINSTEIN J. H., TAYBI H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. (англ.) // American journal of diseases of children (1960). — 1963. — Vol. 105. — P. 588—608. — PMID 13983033.
- ↑ Wójcik C., Volz K., Ranola M., Kitch K., Karim T., O’Neil J., Smith J., Torres-Martinez W. Rubinstein-Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome? (англ.) // American journal of medical genetics. Part A. — 2010. — Vol. 152A, no. 2. — P. 479—483. — doi:10.1002/ajmg.a.33303. — PMID 20101707.
- ↑ Petrij F., Giles R. H., Dauwerse H. G., Saris J. J., Hennekam R. C., Masuno M., Tommerup N., van Ommen G. J., Goodman R. H., Peters D. J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. (англ.) // Nature. — 1995. — Vol. 376, no. 6538. — P. 348—351. — doi:10.1038/376348a0. — PMID 7630403.
- ↑ Исаев Д. Н. Умственная отсталость у детей и подростков. Руководство. — СПб: Речь, 2003. — С. 58. — 397 с. — ISBN 5-9268-0212-1.
Литература[править | править код]
- «Наследственные синдромы и медико-генетическое консультирование», С. И. Козлов, Е. С. Еманова[прояснить]
Источник
Медицинская статья опубликована в рубрике: Педиатрия, СИНДРОМЫ | Декабрь 28th, 2013
 Многопорочный синдром ребенка, описанный Рубинштейном и Тейби в 1963 году, характеризуется постоянным сочетанием аномалий пальцев, черепно-лицевых аномалий, запоздалым развитием веса, роста и психики. Синдром известен и под названием «синдром широких пальцев, с лицевыми аномалиями».
Многопорочный синдром ребенка, описанный Рубинштейном и Тейби в 1963 году, характеризуется постоянным сочетанием аномалий пальцев, черепно-лицевых аномалий, запоздалым развитием веса, роста и психики. Синдром известен и под названием «синдром широких пальцев, с лицевыми аномалиями».
Этиопатогенез синдрома Рубинштейна-Тейби.
Этиопатогенез не установлен. Существование некоторых случаев семейного характера наводит на мысль о генетической причине; передача аномалии могла быть осуществлена доминантным образом или рецессивным, связанным с полом или даже рецессивно — аутосомным. Кариотип, во всех изученных случаях, был нормальным.
Не так давно (1968 — Padfield) выдвинул предположение, которое начинает привлекать все больше сторонников и согласно которому болезнь является вторичной по отношению к заболеванию зародыша (то есть представляет собой эмбриональное заболевание), однако, причина и момент поражения зародыша еще неизвестны.
Синдром Рубинштейна-Тейби встречается очень редко в медицинской практике; начиная с года первого описания до 1970 г. было известно 70 случаев у детей, приблизительно с одной и той же частотой между полами.
Симптоматология синдрома Рубинштейна-Тейби.
Аномалии пальцев: концевые фаланги больших пальцев рук и ног, иногда даже и других пальцев — короткие, широкие и сплющенные, похожие на «шпатель» или «палитру». Это сплющивание относится как к кости, так и к мягкой ткани пальцев. Пальцы короче и толще обычных. Вторая фаланга большого пальца руки перемещается по направлению к лучевой кости, принимая вид подвывиха; большой палец не может быть приведен к ладони.
Другие пальцы расширены на концах, со сплющенными ногтями в форме «чашечек».
Черепно-лицевая дисморфия, проявляющаяся рядом аномалий в самых различных комбинациях, обусловливает характерный вид лица:
- макроцефалия;
- «naevus flacmus» области лба;
- дугообразные брови;
- птоз век;
- экзофтальм;
- гииертелоризм;
- «антимонголоидные» щели век;
- уши внедрены низко со стертыми складками;
- орлиный нос;
- перегородка носа превосходит в нижней части крылья носа;
- микрогнатизм и ретрогнатизм;
- стрельчатое небо.
Запоздалое развитие веса и роста — постоянное клиническое проявление, разной интенсивности. Это в равной мере относится как к росту, так и к весу, реализуя, таким образом, гармоничный нанизм.
Психологическое запоздание постоянно присутствует, но разной интенсивности (умственный дефицит колеблется между 15 и 90% по отношению к норме). Речь очень поздняя и трудная. Аффективность и общительность мало или совсем не изменены.
Изменения папиллярных гребешков (дерматоглифы), хотя и не специфичны, они могут быть показательными: патологическая конфигурация возвышений больших пальцев и малых пальцев и первого межпальцевого пространства; множество извилин большого пальца рук и ног; маленькая ладонная складка.
Диагностика синдрома Рубинштейна-Тейби.
Лабораторные исследования не показывают характерных биогуморальных изменений.
Рентгенологическое исследование выявляет сужение и небольшое утолщение запястья и плюсны, а на пальцах расширение концевых фаланг.
Патологическая анатомия синдрома Рубинштейна-Тейби.
В некоторых случаях, было установлено полное или частичное отсутствие каллезного тела и морфологические аномалии в структуре коры мозга:
- отсутствие больших и средних пирамидальных клеток коры;
- запоздание миелинизации.
Все это показывает на какую-то степень инактивности центральной нервной системы.
Течение и прогноз синдрома Рубинштейна-Тейби: сдержанные, ввиду неврологических проявлений и психического запоздания, иногда очень тяжелого. Все же, чаще всего, больные приспосабливаются к общественной жизни.
Лечение синдрома Рубинштейна-Тейби.
Не существует эффективного специфического лечения.
Источник
Пороки развития орофациальной области: наследственные, мультифакториальные, внутриутробно приобретенные. Аномалии прикуса.
Синдром Рубинштейна-Тейби («синдром широких пальцев, с лицевыми аномалиями». описан в 1963 г.); включает множество дефектов, из которых наиболее частыми являются сочетания интеллектуальной недостаточности (олигофрении) различной степени выраженности с речевыми нарушениями, черепно-лицевыми и пальцевыми аномалиями, Характеризуется постоянным сочетанием аномалий пальцев, черепно-лицевых аномалий, дефектами зрения, запоздалым развитием веса, роста и психики. У мальчиков, кроме того, отмечаются проявления полового недоразвития.
Синдром встречается очень редко в медицинской практике; начиная с года первого описания до 1970 г. было известно 70 случаев у детей, приблизительно с одной и той же частотой между полами.
Существование некоторых случаев семейного характера наводит на мысль о генетической причине; передача аномалии могла быть осуществлена доминантным образом или рецессивным, связанным с полом или даже рецессивно — аутосомным. Кариотип, во всех изученных случаях, был нормальным. При цитогенетическом исследовании в ряде случаев выявляют микроделецию сегмента р13.3 хромосомы 16. Таким образом, тип наследования предположительно аутосомно-доминантный.
Симптоматология синдрома Рубинштейна-Тейби.
1) Аномалии пальцев: концевые фаланги больших пальцев рук и ног, иногда даже и других пальцев — короткие, широкие и сплющенные, похожие на «шпатель» или «палитру». Это сплющивание относится как к кости, так и к мягкой ткани пальцев. Пальцы короче и толще обычных. Вторая фаланга большого пальца руки перемещается по направлению к лучевой кости, принимая вид подвывиха; большой палец не может быть приведен к ладони. Другие пальцы расширены на концах, со сплющенными ногтями в форме «чашечек». Рентгенологическое исследование выявляет сужение и небольшое утолщение запястья и плюсны, а на пальцах расширение концевых фаланг. Возможны многопалость стоп, частичная синдактилия пальцев кистей и стоп;
2) Черепно-лицевые особенности (черепно-лицевая дисморфия):микро– и брахицефалия; выступающий лоб с низким ростом волос; поздно закрывающиеся роднички; ярко-красный невус на коже лба, затылка, шеи; гримаса, напоминающая улыбку; тонкая верхняя губа; смещение верхней челюсти назад; аномалии количества и роста зубов)
-дугообразные, арковидные брови; антимонголоидный разрез глаз – наружные углы глаз расположены ниже внутренних; эпикант; длинные ресницы; косоглазие; птоз век; экзофтальм; гипертелоризм;
-ушные раковины деформированы — внедрены низко и асимметрично, со стертыми складками;
—орлиный нос; перегородка носа превосходит в нижней части крылья носа; широкие переносица и спинка носа; загнутый книзу кончик носа; гипоплазия крыльев носа;
-микрогнатизм и ретрогнатизм; высокое стрельчатое небо; расщелины нёба, язычка, реже – губы;
3) Запоздалое развитие веса и роста — постоянное клиническое проявление, разной интенсивности. Это в равной мере относится как к росту, так и к весу, реализуя, таким образом, гармоничный нанизм. Аномалии грудины и ребер; различные деформации позвоночника; гирсутизм; пороки внутренних органов (дефекты перегородок сердца; аплазия или удвоение почек; гидронефроз; дивертикул мочевого пузыря; крипторхизм) и др. В некоторых случаях, было установлено полное или частичное отсутствие каллезного тела и морфологические аномалии в структуре коры мозга: отсутствие больших и средних пирамидальных клеток коры; запоздание миелинизации. Все это показывает на какую-то степень инактивности центральной нервной системы.
4) Запаздывание психического развития – почти всегда имеется задержка психомоторного и речевого развития (умственный дефицит колеблется между 15 и 90% по отношению к норме). Речь очень поздняя и трудная. Аффективность и общительность мало или совсем не изменены. При данном заболевании дети отличаются благодушным, веселым нравом, они чрезвычайно внушаемы, дружелюбны, направлены на общение с окружающими. В то же время поведение их малоорганизованно, они не критичны, не могут правильно понять ситуацию. Многие из них, особенно мальчики, пугливы, несамостоятельны, и это при импульсивности и возбудимости. Дети с данным заболеванием чрезвычайно чувствительны к одобрению и ласке со стороны взрослых. Это дает возможность родителям сформировать у них социально принятые нормы поведения. Обучению эти дети поддаются с большим трудом даже по специальным программам. Наблюдения за ними в экспериментальной группе показывают, что это связано не только со степенью снижения интеллекта, но и с нарушениями эмоционально-волевой сферы речи, зрения, специфическими трудностями формирования даже элементарных пространственно-временных представлений. Все же чаще больные могут приспосабливаться к общественной жизни.
У больных существует повышенный риск развития опухолей, в основном менингеом и других опухолей мозга, а также лейкемий.
Аномалии прикуса — это отклонение от нормального взаимоотношения зубных рядов верхней и нижней челюстей.
Нормальный прикус характеризуется тем, что верхние резцы слегка перекрывают нижние. Однако идеально ровные от рождения зубы – большая редкость, так что более 80% людей планеты имеют неправильный (аномальный) прикус.
Симптомы неправильного прикуса. Одним из самых очевидных признаков развития аномалии прикуса являются кривые или выпирающие зубы. Возможны как легкие, так и тяжелые формы аномалий прикуса.
Неправильный прикус приводит к увеличению нагрузки на отдельные зубы, что приводит к повышению их чувствительности. Аномалии прикуса обусловливают: трудности с жеванием, постоянное прикусывание щеки или неба, невнятную речь, проблемы с произношением отдельных слов или другие речевые проблемы. Характерна также боль в мышцах лица или челюсти. Если высота прикуса понижается лицо человека может потерять свою симметрию, Явные дефекты прикуса: выступающая вперед нижняя челюсть, оттопырена верхняя губа, кривые зубы.
У взрослых аномалии прикуса обычно остаются такими же, или прогрессируют со временем. У детей симптомы неправильного прикуса могут исчезать со временем, потому что в подростковом возрасте челюсть сильно увеличивается в размере.
Источник

Клинические проявления микроделеционных синдромов

Синдромы с микроделециями Синдром Локализация микроделеции Делеция 1 р36 1 p 36 Вильямса 7 q 11 Лангера-Гидеона 8 q 34 WAGR (опухоль Вильмса, аниридия, пороки 11 р13 мочеполовой системы, задержка роста и развития) Ангельмана и Прадера-Вилли 15 q 11 -q 12 Рубинштейна-Тейби 16 p 13. 3 Миллера-Дикера 17 p 13. 3 Смита-Мажениса 17 p 11. 2 Ди. Джорджи 22 q 11

Ребенок с синдромом делеции 1 p 36: Микроцефалия, задержка роста, умственная отсталость, эпилепсия, гипотония, прямые брови с глубоко посаженными глазами и гипоплазией средней части лица.
 Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций")
Синдромы реципрокных микроделеций/микродупликаций Участок генома (размер) Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций 7 q 11. 23 (1, 5 – 1, 8 Mb) Синдром Вильямса (OMIM 194050) Синдром микродупликации 7 q 11. 23 (OMIM 609757) 17 р11. 2 (~3, 7 Mb) Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) 15 q 11 -q 13 (~4 Mb) Синдром Прадера-Вилли/ Ангельмана (OMIM 17627/ 105830) Синдром микродупликации 15 q 11 -q 13 (OMIM 608636) 17 p 13. 3 (1, 8 -4 Mb) Синдром Миллера-Дикера (OMIM 247200) Синдром микродупликации 17 p 13. 3 (OMIM 613215) 22 q 11. 2 (1, 5 -3 Mb) Синдром Ди Джорджи (OMIM 188400) Синдром микродупликации 22 q 11. 2 (OMIM 608363) Неаллельная гомологичная рекомбинация (NAHR) – механизм рекуррентных микроделеционных/микродупликационных синдромов
 и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат")
Синдром Вильямса (OMIM 194050) и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 126 -66, 924, 597)x 1 Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 12666, 924, 597)x 3

Синдром Вильямса у одного и того же больного в возрастах 7 и 45 лет
 Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем")
Синдром Вильямса (микроделеция 7 q 11) Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем детстве Надклапанный стеноз аорты, стеноз легочной артерии Полные щеки, полная нижняя губа ( «лицо эльфа» ) Покатые плечи «Cocktail – party» манера общения в детстве, замкнутость во взрослом возрасте

микроделеция 7 q 11 Норма Синдром Вильямса
")
Синдром Вильямса (OMIM 194050)


 Специфическое лицо:")
Синдром Лангера-Гидеона (трихоринофалангеальный синдром, микроделеция 8 q 34. 11 -q 34. 13) Специфическое лицо: Грушевидный нос Длинный фильтр Гиперплазия нижней челюсти Тонкая верхняя губа Большие оттопыренные ушные раковины Тонкие волосы

Синдром Лангера-Гидеона Конической формы короткие пальцы Множественные хрящевые экзостозы
 опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и")
WAGR синдром (микроделеция 11 р13) опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и развития
")
Интерстициальная делеция короткого плеча хромосомы 11 (отмечена одной стрелкой)
 Умственная отсталость Аутизм Судороги Насильственный смех")
Синдром Ангельмана (микроделеция 15 q 11 -q 12) Умственная отсталость Аутизм Судороги Насильственный смех Атаксия (синдром «счастливой куклы» ) Брахицефалия Макростомия Увеличение нижней челюсти
 и мозаицизма по микроделеции 15 q")
Сочетание делеции гена CDKL 5 (неонатальная эпилептическая энцефалопатия) и мозаицизма по микроделеции 15 q 11. 2 (синдром Ангельмана) Фенотип: сочетание умственной отсталости, аутизма, микробрахицефалии, приступов смеха, «движений механической куклы» , т. е. клинических признаков эпилептической энцефалопатии, связанной с мутациями гена CDKL 5 и синдрома Ангельмана. Результаты исследования молекулярного кариотипа: arr Хр22. 13(18, 519, 70318, 538, 165)× 1, 11 p 13(35, 140, 75535, 377, 738)× 1, 15 q 11. 2(18, 422, 77021, 062, 213)× 1~2
 Умственная отсталость Выраженная гипотония при рождении")
Синдром Прадера-Вилли (микроделеция 15 q 11 -q 12) Умственная отсталость Выраженная гипотония при рождении и впоследствии Ожирение Гипогонадизм Маленькие кисти и стопы

Синдром Прадера-Вилли Особенности фенотипа: У больных с синдромом Прадера-Вилли вследствие унипарентальной дисомии часто встречаются расстройства аутистического спектра и психозы. Генотип: Сегментная потеря гетерозиготности или унипарентальная дисомия длинного плеча хромосомы 15, локализованная в критическом участке синдромов Ангельмана и Прадера. Вилли. Результаты исследования молекулярного кариотипа: arr 15 q 11. 2(24, 251, 567 – 25, 253, 314)x 2 hmz

Синдром Прадера-Вилли Делеции Унипарентальная изодисомия Синдром Ангельмана

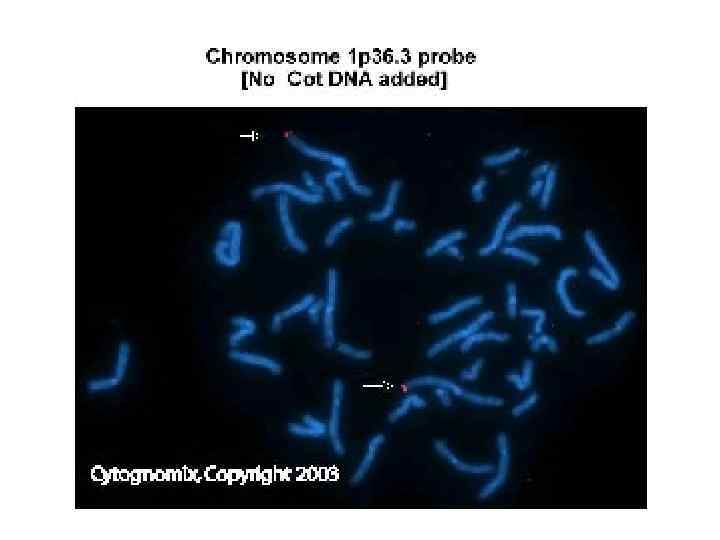
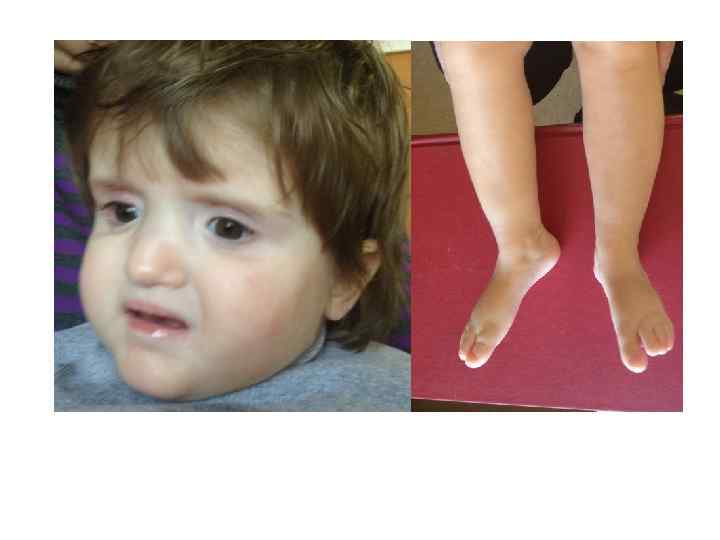
Синдром Рубинштейна-Тейби в 13% случаев микроделеции 16 р13. 3 Специфическое лицо: выступающий лоб, дугообразные брови, антимонголоидный разрез глаз, широкая переносица, эпикант, загнутый вниз кончик носа, напоминающая улыбку гримаса, аномалии роста зубов.

Синдром Рубинштейна-Тейби

Синдром Рубинштейна-Тейби Широкие I пальцы кистей и стоп Умственная отсталость глубокая Отставание в росте (у взрослых рост менее 145 -150 см Микроцефалия Пороки сердца: открытый артериальный проток и дефекты перегородок сердца Пороки почек: односторонняя аплазия, удвоение почек, гидронефроз Пороки мозга: агенезия мозолистого тела Крипторхизм
 Выраженая умственная отсталость, судороги, микроцефалия, высокий")
Синдром Миллера-Дикера (синдром лиссенцефалии, микроделеция 17 р13. 3) Выраженая умственная отсталость, судороги, микроцефалия, высокий лоб, суженный в области висков, выступающий затылок, «карпий» рот, маленькая нижняя челюсть, ротированные назад ушные раковины со сглаженным рисунком. На МРТ отсутствие борозд и извилин в больших полушариях головного мозга (лиссенцефалия – гладкий мозг), недоразвитие серого вещества.
 Особенности лица: Брахицефалия Гипоплазия средней части лица")
Синдром Смита-Мажениса (микроделеция 17 p 11. 2) Особенности лица: Брахицефалия Гипоплазия средней части лица Выступающая нижняя челюсть Широкое плоское лицо Вывернутая верхняя губа Близко посаженные глаза Аномалии зубов

Другие признаки синдрома Смита-Мажениса: Когнитивные нарушения Особенности поведения: склонность к аутоагрессии, нарушения сна Короткие широкие кисти Хриплый низкий голос Непостоянные признаки: Задержка роста Сколиоз Аномалии глаз (микрокорнеа – уменьшение размера роговицы, аномалии радужной оболочки и др. ) Снижение слуха Пороки сердца

Микроделеция 17 p 11. 2 при синдроме Смита -Мажениса
 Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее")
Синдром Смита-Мажениса (OMIM 182290) Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее просыпание, дневная сонливость), импульсивность, агрессия и аутоагрессия, гипотония мышц, походка на широкой основе, нарушения вскармливания, МАР (выпуклый лоб, монголоидный разрез глаз, гипоплазия средней трети лица, короткий нос с открытыми вперед ноздрями, верхняя губа в форме «шатра» , маленькие кисти и стопы, брахидактилия, плоскостопие) Запись результатов исследования молекулярного кариотипа: arr 17 p 11. 2(16, 758, 204 -20, 393, 335)x 1 Более 90% случаев –интерстициальная делеция 17 p 11. 2 размером 3. 7 -Mb, менее 10% — точковые мутации гена RAI 1 внутри данного региона.
 Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического")
Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического спектра, гипотония мышц, открытое овальное окно, гиподонтия, выступающие лобные бугры, гипоплазия нижней челюсти Результат исследования молекулярного кариотипа: 17 p 11. 2(16, 458, 870 -16, 350)x 3
")
Синдром Ди. Джорджи (делеция 22 q 11)

Синдром Ди-Джорджи • Гипо- или аплазия тимуса, ведущая к нарушениям иммунитета и генералированным инфекциям • Гипоплазия паращитовидных желез, ведущая к гипокальциемическим судорогам у новорожденных • Пороки сердца (тетрада Фалло). • Особенности лица: гипертелоризм или телекант, антимонголоидный разрез глаз, укороченный фильтр, маленькая нижняя челюсть, низко расположенные ушные раковины

Метод молекулярного кариотипирования – прорыв в исследовании микроделеционных синдромов Лаборатория молекулярной цитогенетики НИИ педиатрии и детской хирургии нервно -психических заболеваний Лаборатория цитогенетики и и геномики психических заболеваний Научного центра психического здоровья
 Фенотип: Задержка психомоторного и речевого развития, частые")
Синдром микродупликации гена MECP 2 (OMIM 300260) Фенотип: Задержка психомоторного и речевого развития, частые респираторные инфекции, крипторхизм, микробрахицефалия, широкое лицо, эпикант, гипоплазия средней части лица, заостренный нос, маленький полуоткрытый рот, крупные низко расположенные ушные раковины Генотип пробанда: arr Xq 28(153, 130, 000– 153, 647, 227)x 3

Сравнение клинических признаков у 4 больных с дупликациями Xq 28, включающими ген MECP 2, с ранее описанными случаями Признаки, наблюдавшиеся у Случай 1 Случай 3 Случай Частота 2 детей с дупликациями Xq 28 4 признака (%) по данным литературы Задержка физического развития — + + + 51 Микроцефалия + + 13 Рекуррентные инфекции + + — + + 83 Эпикант + + Крупные ушные раковины + + 20 Маленький рот + + 30 Широкое лицо + + — + 50 Гипоплазия гениталий/ крипторхизм Судороги + + 50 — + + + 52 Гипотония мышц конечностей и лицевой мускулатуры Отсутствие речи + + 68 + + 62 Задержка психомоторного развития + — + + + 100 + 52 Проблемы со вскармливанием 13

Открытие этиологии ранее описанных синдромов
 Задержка психического развития,")
Синдром субтеломерной микроделеции 9 q 34. 3 (Kleefstra syndrome, OMIM 610253) Задержка психического развития, гипотония мышц и особенности лица (микробрахицефалия, синофриз, необычная форма бровей, гипоплазия средней части лица, вывернутая нижняя губа) Более 85% случаев заболевания – микроделеция 9 q 34. 3. Менее 15% — мутации в гене EHMT 1
Синдром субтеломерной микроделеции 9 q 34. 3 (Kleefstra syndrome, OMIM 610253) Фенотип: Задержка психического развития, аутистические черты в поведении, гипотония мышц, особенности лица (микробрахицефалия, синофриз, гипоплазия средней части лица, открытые вперед ноздри, вывернутые губы). Аномалии строения мозга – корковая лобная атрофия, вентрикуломегалия. Врожденный порок сердца – клапанный стеноз аорты, двустворчатый аортальный клапан, клапанный стеноз легочной артерии Результаты исследования молекулярного кариотипа: arr 9 q 34. 3(139, 925, 932 141, 020, 389)x 1 Размер: 1, 474, 096 пн 62 гена Более 85% случаев заболевания – микроделеция 9 q 34. 3. Менее 15% — мутации в гене EHMT 1
 Фенотип: Умственная отсталость, судороги Приступы гипервентиляции Хронические запоры (болезнь Гиршпрунга)")
Синдром Питта-Хопкинса (OMIM 610954) Фенотип: Умственная отсталость, судороги Приступы гипервентиляции Хронические запоры (болезнь Гиршпрунга) Специфическое лицо (выступающие надбровные дуги, широкий уплощенный нос, большой рот с изогнутой верхней губой) Кариотип 46, XX, der(18) Запись результатов исследования молекулярного кариотипа: arr 2 q 24. 2(160, 185, 503 -162, 242, 742)× 1, 16 q 22. 1(68, 649, 502 -68, 749, 192)× 1, 18 q 21. 2 q 21. 32 (50, 396, 511 -57, 063, 895)× 1 Синдром клинически описан Pitt и Hopkins в 1978 г. Микроделеция 18 q 21. 2 (1, 2 и 1, 8 Mb) – 2 случая Точковые мутации в гене TCF 4 описаны Zweier et al. (2007) ,

Диагностика моногенных заболеваний

Микроделеции, ключевые симптомы которых – результат гаплонедостаточности одного гена Синдром Моуат-Вильсона, OMIM 235730 Фенотип: Умственная отсталость, микроцефалия, эпилепсия, низкий рост, врожденные аномалии сердца, болезнь Гиршпрунга, агенезия мозолистого тела на МРТ, характерное лицо (гипертелоризм, сходящееся косоглазие, закругленный кончик носа, широкая носовая перегородка, постоянно открытый рот, заостренный подбородок, ротированные назад ушные раковины) Результаты исследования молекулярного кариотипа: arr 2 q 22. 1 q 23. 3 (140, 410, 739 -150, 635, 360)x 1 Размер: 10, 224, 621 56 генов, 13 – в OMIM Синдром клинически описан Mowat в 1998 г. Описаны более 180 случаев точковых мутаций в гене