Саркома позвоночника код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Саркома позвоночника.
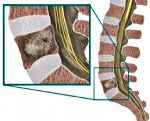
Саркома позвоночника
Описание
Саркомы позвоночника. Группа злокачественных опухолей, происходящих из незрелой соединительной ткани. Могут поражать кости, хрящи, сосуды и другие соединительнотканные структуры позвоночника. Проявляются болями, тазовыми расстройствами, нарушениями чувствительности и движений. Клиническая картина определяется видом саркомы позвоночника, уровнем поражения, расположением опухоли относительно спинного мозга и степенью повреждения спинного мозга. Диагноз выставляется с учетом анамнеза, жалоб, данных осмотра, результатов рентгенографии, КТ, МРТ, биопсии и других исследований. Лечение – операция, химиотерапия, радиотерапия.
Дополнительные факты
Саркомы позвоночника – группа редко встречающихся соединительнотканных злокачественных опухолей позвоночника, характеризующихся взрывным ростом и быстрым прогрессированием. Несмотря на незначительную долю в общей структуре онкологической заболеваемости, саркомы позвоночника являются второй после рака причиной смертности больных злокачественными новообразованиями. Причиной большого количества летальных исходов становятся инфильтративный рост, раннее метастазирование и частое рецидивирование. Саркомы позвоночника особенно агрессивно протекают у детей и подростков, что обусловлено быстрым ростом соединительнотканных структур в этом возрастном периоде. Чаще страдают лица мужского пола. Лечение осуществляют специалисты в области онкологии, вертебрологии и неврологии.
Саркома позвоночника
Причины
Причины развития саркомы позвоночника точно не установлены. Предполагается, что заболевание возникает под влиянием группы факторов, в число которых входят некоторые ДНК- и РНК-содержащие вирусы, ионизирующее облучение, контакт с канцерогенными веществами Ученые отмечают, что саркома позвоночника чаще развивается у лиц, в прошлом перенесших переломы и ушибы позвоночного столба. При этом большинство исследователей считают, что травма не является непосредственной причиной развития саркомы позвоночника, а лишь провоцирует рост уже имеющихся в организме аномальных клеток.
Существуют научные данные в пользу теории неблагоприятной наследственности. У пациентов детского и юношеского возраста имеет значение интенсивный рост костной, хрящевой и мышечной ткани. Установлено, что остеосаркома и саркома Юинга обычно развиваются у детей и подростков и крайне редко встречаются у больных старше 30-35 лет. Вместе с тем, некоторые виды сарком позвоночника поражают преимущественно людей старше 30 лет. Предполагается, что в подобных случаях определенное значение могут иметь некоторые заболевания позвоночника. При развитии сарком позвоночника у людей старше 40-45 лет обычно предполагают малигнизацию доброкачественной опухоли либо развитие болезни на фоне воспалительного или дегенеративно-дистрофического процесса.
Классификация
С учетом происхождения выделяют следующие виды сарком позвоночника:
• Остеосаркома – происходит непосредственно из костной ткани.
• Хондросаркома – происходит из хрящевой ткани.
• Саркома Юинга – происходит из ретикулоэндотелиальной ткани костного мозга.
• Ангиосаркома – происходит из ткани сосудов, обеспечивающих кровоснабжение позвоночного столба.
• Периостальная фибросаркома – происходит из наружного слоя надкостницы.
Наряду с перечисленными саркомами позвоночника, к онкологическим поражениям соединительной ткани позвоночного столба можно отнести миеломную болезнь – близкую к лейкозам злокачественную опухоль из плазматических клеток, очаги которой нередко обнаруживаются в позвонках. Кроме того, в позвоночнике могут выявляться узлы, возникшие в результате метастазирования сарком других локализаций. Такие новообразования называются метастатическими саркомами.
С учетом уровня дифференцировки клеток различают три вида сарком позвоночника: высокодифференцированные, среднедифференцированные и низкодифференцированные. Низкодифференцированные саркомы позвоночника быстрее растут, раньше метастазируют и чаще рецидивируют. Чем ниже уровень дифференцировки – тем более неблагоприятен прогноз. С учетом уровня поражения выделяют опухоли шейного, грудного, поясничного, крестцового и копчикового отдела позвоночника, с учетом локализации – новообразования, расположенные по передней, задней и боковой поверхности спинного мозга.
Симптомы
Первым симптомом заболевания обычно становится боль. Вначале болевой синдром выражен слабо или умеренно, боли носят интермиттирующий характер, обычно усиливаются в ночное время. В отличие от болевого синдрома, обусловленного дегенеративно-дистрофическими процессами, боли при саркоме позвоночника не уменьшаются в покое. Из-за стертой симптоматики, отсутствия онкологической настороженности в отношении молодых пациентов и неправильной трактовки проявлений онкологического процесса (например, предположений о травматическом генезе болевого синдрома из-за частых незначительных травм у детей и подростков) на ранних стадиях саркома позвоночника нередко остается нераспознанной.
При прогрессировании интенсивность болей нарастает. Пациенты не могут заснуть или просыпаются от боли по ночам. Подвижность позвоночника ограничивается. Возникает корешковый синдром (радикулит), в зависимости от уровня поражения больные саркомой позвоночника предъявляют жалобы на боли в руках, ногах, пояснице или внутренних органах. При сдавлении спинного мозга вначале возникают нарушения чувствительности и движений, затем развиваются парезы, параличи и тазовые расстройства. У всех пациентов, страдающих саркомой позвоночника, отмечается снижение трудоспособности и нарушение повседневной активности. Выявляются анемия, повышение температуры, слабость, апатия, ухудшение аппетита и снижение веса. На поздних стадиях саркомы позвоночника возникают патологические переломы. Наблюдается гематогенное метастазирование с поражением легких, костей и головного мозга. Другие органы страдают редко.
Хондросаркома позвоночника также является редкой злокачественной опухолью и составляет около 2,5% от общего количества хондросарком. Может быть первичной (возникшей из неизмененного хряща) или вторичной (развившейся на фоне хондроматоза, хондромы, хондромиксоидной фибромы или хондробластомы). Эта разновидность саркомы позвоночника диагностируется у лиц старше 30 лет, в детском возрасте встречается редко. Мужчины страдают чаще женщин. У молодых больных отмечается более бурное течение и быстрое прогрессирование. Поражаются преимущественно крестец и поясничные позвонки.
Саркома Юинга является одной из наиболее агрессивных злокачественных опухолей. На момент постановки диагноза отдаленные метастазы выявляются у 14-50% пациентов. Этот вид саркомы позвоночника развивается в детском и юношеском возрасте, после 30-35 лет встречается крайне редко. Мужчины страдают чаще женщин. Первичная опухоль представляет собой одиночный узел, обычно расположенный в теле позвонка. При прогрессировании саркома позвоночника может прорастать близлежащие структуры позвоночного столба, давать метастазы в другие позвонки и в ткань легких. Лимфатические узлы и другие кости скелета страдают редко.
Ангиосаркома. Злокачественная опухоль сосудистого происхождения. Прогрессирует быстрее доброкачественных сосудистых опухолей, разрушает костные структуры и прорастает окружающие мягкие ткани. Эта разновидность саркомы позвоночника вызывает выраженные неврологические нарушения, часто становится причиной патологических переломов позвонков.
Боль в шейном отделе позвоночника. Изменение аппетита. Изменение веса. Истощение. Ломота в мышцах. Нарушение терморегуляции. Отсутствие аппетита. Потеря веса. Слабость.
Диагностика
Диагноз устанавливается с учетом жалоб, анамнеза, результатов общего и неврологического осмотра, данных инструментальных и лабораторных исследований. При проведении рентгенографии у больных саркомой позвоночника в пораженном позвонке обнаруживается очаг деструкции с неровными контурами. Выявляется разрушение кортикального слоя и округлая тень мягкотканного компонента новообразования. КТ и МРТ позвоночника позволяют уточнить размеры опухоли и ее взаимоотношения с близлежащими анатомическими структурами. В некоторых случаях для уточнения диагноза назначают сцинтиграфию.
Наряду с перечисленными методиками в процессе диагностики саркомы позвоночника используют исследование крови на онкомаркеры, иммуногистохимические и молекулярно-генетические исследования. Окончательный диагноз выставляют на основании результатов гистологического исследования. Забор материала осуществляют путем пункционной биопсии позвонка под контролем КТ или рентгеноскопии. Для выявления отдаленных метастазов назначают рентгенографию грудной клетки, КТ и МРТ головного мозга, рентгенографию и сцинтиграфию всего скелета.
Лечение
Радикальное оперативное удаление опухоли зачастую невозможно из-за прорастания близлежащих тканей. При компрессии спинного мозга осуществляют паллиативные хирургические вмешательства. Ведущую роль в лечении саркомы позвоночника отводят комбинированной терапии. Назначают радиотерапию и полихимиотерапию с использованием этопозида, циклофосфана, доксорубицина и других препаратов. При отдаленных метастазах для увеличения эффективности лечения применяют лучевую терапию в высоких дозах с последующей пересадкой костного мозга или введением стволовых клеток.
Прогноз
Прогноз зависит от стадии процесса, распространенности и уровня дифференцировки саркомы позвоночника. Использование комбинированной терапии при локальных высокодифференцированных опухолях обеспечивает устойчивую ремиссию у 60-70% пациентов. Высокодозное облучение с последующей трансплантацией костного мозга позволяет добиться излечения 30% больных. При высокой чувствительности саркомы позвоночника к химио- и радиотерапии вероятность благоприятного исхода возрастает, половине пациентов удается дожить до 7 лет с момента окончания лечения.
Источник
Саркома Юинга (лат. myeloma endotheliale) — злокачественная опухоль костного скелета. Саркома Юинга, как правило, поражает нижнюю часть длинных трубчатых костей, ребра, таз, лопатку, позвоночник и ключицу.
Была открыта Джеймсом Юингом (1866—1943) в 1921 году. Учёный охарактеризовал её как опухоль, поражающую в основном длинные трубчатые кости.
Саркома Юинга является одной из самых агрессивных злокачественных опухолей [1]. До применения системной терапии почти у 90 % больных развивались метастазы. Наиболее частая локализация метастазов на момент первичной диагностики — лёгкие, кости, костный мозг. 14—50 % пациентов к моменту установления диагноза уже имеют метастазы, выявляемые рутинными методами исследования, и гораздо больше больных имеют микрометастазы. Лимфогенное распространение метастазов встречается редко и всегда связано с плохим прогнозом. Редко также имеет место ретроперитонеальное и медиастинальное распространение метастазов. 2,2 % пациентов имеют метастазы в ЦНС при первичной диагностике, и почти все — при генерализации процесса.
Описание заболевания[править | править код]
Распространение заболевания[править | править код]
Для костных сарком характерен быстрый рост и раннее метастазирование. Саркома Юинга является второй по частоте среди злокачественных опухолей костей у детей — составляет 10-15 %. Эта опухоль редко встречается у детей моложе 5 лет и у взрослых старше 30 лет. Пик заболеваемости приходится на 10 −15 лет.
Заболеваемость саркомой Юинга имеет отчетливые географические и этнические особенности. Значительно чаще опухоли этой группы регистрируются у белых подростков по сравнению с жителями стран Африки и Азии. Различия в заболеваемости саркомой Юинга по
половому признаку появляются после достижения больными возраста 13-14 лет. Мальчики болеют чаще, чем девочки в соотношении приблизительно 1,5:1.
В возрасте до 20 лет саркомой Юинга чаще поражаются длинные (бедренная, мало- и большеберцовые, плечевая) кости, в более старшем возрасте – плоские кости таза и черепа, ребра, лопатки, позвонки.
Не менее 70% всех сарком Юинга локализуются на нижних конечностях и в области тазового пояса. Первичное вовлечение в опухолевый процесс костей верхних конечностей, в первую очередь плечевой кости, составляет от 12 до 16%. Более редкими, не превышающими по частоте 10-13%, локализациями саркомы Юинга являются позвонки, ребра, ключицы, лопатки, кости черепа, мелкие кости кисти и стопы.
Причины возникновения[править | править код]
К настоящему времени не удалось выявить потенциальные причины возникновения саркомы Юинга. Существует ряд научных данных, свидетельствующих о роли наследственного компонента в механизме развития заболевания. В частности, описано одновременное развитие
саркомы у сиблингов, что позволяет судить о значении генетических дефектов. Также доказано, что в 40 % случаях возникновение костной саркомы провоцирует травма.[источник не указан 3131 день]
Саркома Юинга состоит из мелких круглых клеток со скудной цитоплазмой, круглым ядром, содержащим нежный хроматин и слабо просматривающиеся базофильные нуклеолы. В отличие от остеосаркомы, она не продуцирует остеоид.
Идея эндотелиальной природы опухоли Юинга превалировала до 1980 года. Исследования, проведенные в последние годы, показали нейрогенную природу опухоли Юинга. Чаще саркома Юинга является недифференцированной опухолью костей. В специальной литературе появился термин «семейство опухолей типа саркомы Юинга». К нему относят: собственно саркома Юинга; периферические примитивные нейроэктодермальные опухоли (PNET), в том числе PNET костей, и экстраоссальная саркома Юинга.[2]
Клиническая картина[править | править код]
Общие симптомы[править | править код]
Сложность ранней диагностики объясняется отсутствием онкологической настороженности в отношении лиц молодого возраста, стертой клинической симптоматикой, неправильной интерпретацией жалоб больного (например, объяснением их возникновения спортивной или бытовой травмой и т.п.).
- Боль (от момента появления первых болевых ощущений до установления диагноза проходит от 6 до 12 месяцев)
- Первоначально:
- слабая и умеренной интенсивности
- интермиттирующий характер — может самопроизвольно ослабевать и даже полностью купироваться (т.н. «светлые промежутки»)
- не стихает в покое
- усиление по ночам
- отсутствует облегчение при фиксации конечности.
- По мере роста опухоли боль:
- становится интенсивнее
- ограничивает движения в близлежащем суставе вплоть до контрактуры
- нарушает сон
- нарушает повседневную активность
- Первоначально:
- Быстроувеличивающаяся опухоль нередко с патологическим переломом (поздний признак — 3-4 мес)
- Признаки местного воспаления:
- болезненность при пальпации
- гиперемия кожи
- пастозность (отечность) кожи
- локальное повышение температуры
- расширенные подкожные вены
- Синдром общей опухолевой интоксикации
- повышение температуры тела больного (субфебрильная и фебрильная лихорадка)
- снижение веса и аппетита вплоть до кахексии
- слабость
- анемия
- регионарный лимфаденит
- Метастазы
- в лёгочную ткань (чаще всего)
- в костную ткань
- в костный мозг
- отдаленные метастазы (очень редко): в висцеральных органах, лимфатических узлах средостения и забрюшинного пространства, плевре, центральной нервной системе в виде поражения менингеальных оболочек и вещества головного и спинного мозга.
В зависимости от локализации[править | править код]
- При поражении нижней конечности — хромота
- При поражении позвонков:
- радикулопатия
- компрессионно-ишемическая миелопатия с явлениями параплегии,
- нарушением функции тазовых органов (недержание мочи)
- При поражении костей и мягких тканей грудной стенки (в зарубежной литературе носит название опухоли Аскина):
- плевральный выпот
- дыхательная недостаточность
- кровохарканье
Диагностика[править | править код]
- Рентгенография костей, пораженных опухолью и её метастазами
- Рентгенография и КТ легких
- КТ или МРТ участков скелета, мягких тканей и любых других анатомических областей, пораженных опухолевым процессом. Наиболее точно определяет размеры опухоли, её связь с окружающими тканями, сосудисто-нервным пучком, распространение опухоли по костно-мозговому каналу.
- Позитронно-эмиссионная томография (ПЭТ)
- Остеосцинтиграфия. Позволяет диагностировать отдаленные метастазы
- Ангиография
- УЗИ
- Исследование костного мозга (билатеральная трепано-биопсия костного мозга из крыльев подвздошных костей). Особенностью опухолевого процесса саркомы Юинга является изолированное поражение костного мозга при отсутствии метастазов в костях вне зависимости от первичной локализации или размера опухоли
- Биопсия опухоли. Материал получают из участка кости, граничащего с костномозговым каналом или мягкотканного компонента
- Иммуногистохимическое исследование. Практически 100% клеток саркомы Юинга вырабатывают (экспрессируют) на своих мембранах поверхностный гликопротеин СD99 (р30/32MIC2). Определение его экспрессии является убедительным подтверждением клинико-рентгенологического диагноза саркомы Юинга.[3] Кроме того, для клеток саркомы Юинга характерна экспрессия виментина.
- Молекулярно-генетическое исследование.
- Флуоресцентная гибридизация in situ. Практически 90-95% опухолевых клеток имеют транслокацию между 11-й и 22-й хромосомами (t (11;22) (q24;q12)), приводящую к синтезу патологического белка EWS/FLI1 [4]. Определение данного генетического дефекта является патогномоничным симптомом (характерным только для этого заболевания) саркомы Юинга.
- Полимеразная цепная реакция (RT-PCR). Более чувствительный метод. Используется для определения микрометастазов саркомы Юинга в костном мозге и периферической крови.
Гистологическая картина[править | править код]
Классическая гистологическая картина саркомы Юинга представлена бесструктурными агрегатами мелких опухолевых клеток, разделенных фиброзными прослойками. Клетки имеют правильную форму, содержат округлые или овальные ядра, характеризуются высоким ядерно-цитоплазматическим отношением. Заключенная в клеточных ядрах дисперсия хроматина придает им характерный «зеркальный» вид. Митотическая активность в клетках саркомы Юинга, как правило, низкая. Зачастую при патоморфологическом исследовании определяется выраженный некроз опухолевой ткани, с преимущественной локализацией жизнеспособных клеток вокруг сосудов. Особенности морфологии саркомы Юинга затрудняют проведение дифференциальной диагностики с другими мелкоклеточными злокачественными опухолями у детей (нейробластомой, рабдомиосаркомой, неходжкинской лимфомой, синовиальная саркома, лейомиосаркома и др.). Поэтому проведение биопсии недостаточно для постановки диагноза саркомы Юинга [5]
Рентгенологическая картина[править | править код]
- Сосуществование деструктивного и реактивного (остеосклеротического) процессов костеобразования.
- Распространение опухолевого процесса на кортикальный слой вызывает его разволокнение, расслоение, нечеткость контуров, секвестрацию.
- Вовлечение надкостницы стимулирует периостальное костеобразование пластинчатого или игольчатого типа.
- Практически всегда имеется мягкотканный компонент опухоли, по размерам нередко превосходящий участок первичной костной деструкции.
- Характерна однородная структура мягкотканного опухолевого компонента, без элементов патологического костеобразования, обызвествлений или хрящевых включений
Магнитно-резонансная томография[править | править код]
- метод выбора при стадировании опухоли
- оценка ответа на химио- лучевую терапию
- позволяет оценить поражение мягких тканей
- низкий сигнал на Т1 взвешенных изображениях
- гетерогенное контрастное усиление
- гетерогенно высокий МР сигнал на Т2 взвешенных изображениях
Лечение[править | править код]
- Многокомпонентная химиотерапия (используются препараты — винкристин, адриамицин, ифосфамид, циклофосфан, актиномицин, вепезид в комбинации). В современных программах лечения применяется предоперационная и послеоперационная полихимиотерапия, при этом учитывается также гистологический ответ опухоли на лечение. Хорошим ответом опухоли на химиотерапию считается наличие менее 5 % живых опухолевых клеток[источник не указан 3131 день].
- Лучевая терапия на очаг в высоких дозах. При развитии метастазов в лёгкие проводится лучевая терапия на лёгкие.
- Если возможно, радикальное удаление опухоли (включая кость и мягкотканный компонент). Радикальная резекция возможна при очаге в малоберцовой кости, костях предплечья, ребрах, ключице, лопатке.
Операция улучшает локальный контроль опухоли. В сочетании с интенсивной химиотерапией и лучевой терапией значительно снижается риск местного рецидива. Уменьшение частоты местного рецидива отмечается даже после нерадикальных операций. Современная хирургическая техника позволяет проводить органосохраняющие операции при поражении бедренной, плечевой костей, а также резекцию костей таза.
Пациентам с плохим прогнозом, в частности с метастазами в кости и костный мозг, имеющим выживаемость менее 10 %, в последнее время назначают более интенсивное лечение — химиотерапия мегадозами препаратов с тотальным облучением тела и трансплантацией аутологичного костного мозга или периферических стволовых клеток. Эта терапия позволяет излечить более 30 % больных с распространенным процессом (при метастазах в кости и костный мозг). У больных с хорошей чувствительностью опухоли удается достичь ещё более высоких результатов лечения (7-летняя выживаемость составляет около 50 %).
Литература[править | править код]
- О саркоме Юинга на medline.ru
- Саркома Юинга (недоступная ссылка) Пер. с англ. Н. Д. Фирсовой (2017)
- Саркома Юинга у детей
См. также[править | править код]
- Карцинома
- USP6
Ссылки[править | править код]
- Сайт Противоракового Общества России
- Radiographia
Примечания[править | править код]
- ↑ Medline.ru — Биомедицинский журнал
- ↑ Архивированная копия (недоступная ссылка). Дата обращения 30 сентября 2012. Архивировано 3 сентября 2013 года.
- ↑ Kovar H., Dworzak M., Strehl S. et al. Overexpression of the pseudoautosomal gene MIC2 in Ewing’s sarcoma and primitive neutoectodermal tumor // Oncogene. – 1990. – Vol.5. – P.1067.
- ↑ de Alva E., Kawai A., Healey J.H. et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma // J. Clin. Oncol. – 1998. – Vol.16. – P.1248.
- ↑ Fletcher C.D., Unni K.K., Mertens F. Pathology and genetics of tumors of soft tissue and bone. – Lyon: IARC Press, 2002.
Источник