Рото лице пальцевой синдром фото

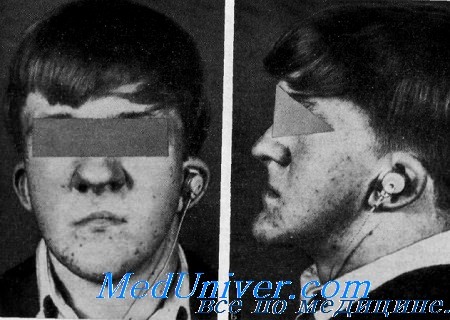
Синдром Мора. Рото-лице-пальцевой или OFD II синдромСиндром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа, гипоплазией тела нижней челюсти полидактилией и синдактилией, а также проводящей глухотой, был описан Mohr и Claussen у 4 из 7 сибсов, а также Rimoin и Edgerton у 3 из 4 сибсов. Rimoin и Edgcrton разделили ротолице-пальцевой синдром на две самостоятельные, генетически различные нозологические формы. Одна из них наследуется Х-сцепленным: доминантным путем (OFD I), а другая — аутосомно-рецессивным путем (OFDII). Gorlin, Pindborg и Cohen представили обзор всех опубликованных случаев синдрома. Клинические данные. Данные осмотра. При характеристике лица отмечают значительную гипоплазию скуловых дуг, верхней челюсти и тела нижней челюсти. Переносье широкое и уплощенное. Часто наблюдается срединное расщепление верхней губы, иногда наблюдается расщепление неба и языка. Описаны также жировые гамартомы и общая неподвижность языка. Постоянным симптомом является двусторонняя ульнарная шестипалость на руках и двусторонняя полисиндактилия I пальцев. В немногих случаях отмечается пять пальцев с отклонением V пальца в сторону локтевой кости, синдактилией III и IV пальцев с дополнительными косточками в соединительной ткани пли с шестипалостью только на руках (Gorlin et al.). Орган слуха. Один из 3 сибсов, описанных Rimoin и Edgerton, был мертворожденным; у 2 остальных была выявлена умеренная двусторонняя проводящая глухота. Другие аудиометрические тесты не описаны. У одного из сибсов при помощи тимпанотомии были выявлены врожденное уродство наковальни и недостаточность сустава между наковальней и стременем. Длинный отросток наковальни имел вид «тупой колбасы», а лентовидный отросток вообще отсутствовал. У его сестры также наблюдалась умеренная проводящая глухота. Тимпанотомия не производилась. Goldstein и Medina сообщили, что у обоих сибсов была проводящая глухота с 40% потерей слуха.
Лабораторные данные. Рентгенограммы показали гипоплазию скуловых дуг и тела нижней челюсти. Остальные костные изменения, выявленные главным образом в конечностях (руках и ногах), так же как полидактилия и синдактилия, обсуждены выше. Наследственность. В семье, описанной Mohr и Claussen, были больны 4 брата. Rimoin и Edgerton обнаружили среди 4 сибсов 3 больных. Goldstein и Medina выявили синдром у 2 из 3 сибсов. Некоторые другие примеры выявления синдрома у сибсов опубликовали Gorlin с сотр.. Так как родители во всех случаях были здоровы, аутосомно-рецессивпое наследование очевидно. Диагноз. OFD I синдром характеризуется гипоплазией крыльев носа, преходящей сыпью па лице, тонкими извитыми волосами, нормальным слухом и Х-сцепленным доминантным наследованием с летальностью для мальчиков (Gorlin et al.). Лечение. Расщепление губы, неба и языка может быть восстановлено хирургическим путем. Лишний палец может быть удален. Глухоту также можно лечить хирургическими методами, вставлением протеза вместо аномальных слуховых косточек. Можно также использовать слуховые аппараты. Прогноз. Глухота является врожденной и не прогрессирует. Хотя у большинства больных продолжительность жизни нормальная, некоторые дети умирают от респираторных инфекций (Gorlin et al.). Выводы. Синдром OFD II характеризуется: 1) аутосомно-рецессивным наследованием; 2) аномалиями лица с гипоплазией тела нижней челюсти, уплощением переносья и широко расставленными медиальными углами глаз; 3) аномалиями пальцев, включая полидактилию, синдактилию и брахидактилию; 4) дольчатый язык; 5) проводящую глухоту вследствие уродства слуховых косточек. — Также рекомендуем «Дисплазия эпифиза головки бедра, близорукость и глухота» Оглавление темы «Наследственная глухота»:
|
Источник
Врождённые пороки челюстно-лицевой области (включая расщелины лица и синдромы первой-второй жаберных дуг)
Врожденные пороки развития головы, лица и шеи целесообразно разделить на следующие группы:
• изолированные врожденные пороки (например, врожденная расщелина губы и/или неба);
• челюстно-лицевые дизостозы (например, синдром Гольденхара, синдром гемифациальной микросомии), когда имеются аномалии развития мягких тканей, костей лицевого скелета, челюстей, однотипно выраженные, что и определяет их как синдром;
• черепно-челюстно-лицевые дизостозы (например, синдром Крузона, синдром Робена), когда имеются врожденные аномалии развития костей черепа, лицевых костей, мягких тканей, также с повторяющимися признаками в однотипной комбинации.
СИНДРОМЫ КРАНИОФАЦИАЛЬНОЙ МИКРОСОМИИ
К синдромам первой и второй жаберных дуг (они же – синдромы краниофациальной микросомии) относятся:
• синдром гемифациальной микросомии,
• синдром Гольдерхара,
• синдром Тричера-Коллинза-Франческетти.
В настоящее время используется классификация OMENS-plus.
К основным симптомам синдрома краниофациальной микросомии относятся:
• изменение размера и положения орбиты (О),
• недоразвитие и деформация нижней челюсти (М),
• недоразвитее, деформация, изменение положения ушной раковины, частичная или полная атрезия наружного слухового прохода (E),
• парез одной или нескольких ветвей лицевого нерва (N),
• недоразвитие мягких тканей лица (S).
Вспомогательные симптомы:
• наличие поперечных, косых расщелин лица, расщелин нёба, губы, альвеолярного отростка,
• наличие эпибульбарных липодермоидов,
• деформация шейных позвонков,
• гемипарез мягкого нёба.
Помимо деформации самой ушной раковины, в зависимости от степени выраженности симптома, могут иметься следующие нарушения:
• предушные придатки,
• предушные свищи, которые при микротии могут как оканчиваться слепо на любом из участков рудимента или открываться в глотку,
• снижение линии роста волос,
• нарушение и определённые особенности кровоснабжения и иннервации височной области,
• недоразвитие височно-теменного листка фасции,
• недоразвитие височной мышцы,
• недоразвитие среднего и внутреннего уха,
• сглаженность или отсутствие заушной борозды,
• вдавленность чешуи височной кости,
• уменьшение пневматизации сосцевидного отростка височной кости.
Лечение синдромов краниофациальной микросомии проводится последовательно и поэтапно. Сроки реабилитации зависят от степени выраженности каждого из симптомов.
При наличии предушный придатков и поперечных расщелин лица, они устраняются в период новорожденности.
При наличии выраженных дефектом и деформаций челюстных костей проводится замещение дефекта эндопротезом или компрессионно-дистракционный остеогенез в возрасте, начиная с 5 лет (по показаниям). При незначительных деформациях коррекция прикуса и несимметричных деформаций откладывается до постпубертатного периода (после 15 лет).
При тотальной аплазии ветви нижней челюсти, с целью нормализации прикуса, профилактики усугубления деформации, создания дистальной опоры нижней челюсти, нормализации костно-мышечного равновесия, используется костная пластик адефекта, или эндопротезирование. Эндопротезирование проводится в любом возрасте, костная пластика — с 12-14 лет.
Реконструкция ушной раковины при её аплазии или значительной деформации осуществляется в среднем в 8-10 лет (в зависимости от физического развития ребёнка). В случае наличия выраженной костной деформации и значительного недоразвития мягких тканей отопластика проводится после устранения данных симптомов. При субтотальных дефектах и незначительных деформациях, а также деформациях по типу «свёртуного уха» — после 6 лет.
Подробнее: https://childrensurgery.ru/rekonstrukciya-ushnoy-rakoviny
При выраженном дефиците мягких тканей проводится пластика лоскутами на сосудистых анастомозах, а также ротационными лоскутами. При незначительной асимметрии в постпубертатном периоде (после 15 лет) проводится липофилинг.
Ортогнатические операции (по исправлению прикуса и окклюзии) и контурная пластика проводится после завершения других этапов лечения.
ПРОЧИЕ СИНДРОМЫ
Синдром Ван-дер-Вуда — врожденные симметричные свищи слизистых желез на нижней губе в сочетании с расщелиной верхней губы (чаще с двусторонней полной расщелиной губы, альвеолярного отростка и неба). Наследуется по аутосомно — доминантному типу с высоким уровнем риска последующего рождения ребенка с подобной патологией и, как правило, более выраженными признаками ее проявления.
Синдром Робена характеризуется наличием триады признаков: расщелины неба, недоразвития продольных размеров нижней челюсти, птоза языка и глотательных мышц за счет врожденного несовершенства функций черепных нервов. Большинство детей ранее были нежизнеспособны из-за нарушения дыхания и развития бронхо — легочных осложнений с момента рождения. В настоящее время посредством технологии дистракционного остеогенеза эти осложнения могут быть своевременно устранены или минимизированы за счет увеличения продольных размеров нижней челюсти, что изменяет положение корня языка, увеличивает площадь дна полости рта и снимает основные условия порочного влияния на функцию дыхания.
Синдром Крузона — изменение формы мозгового черепа («башенный» череп), высокое переносье с выдающимся вперед носом, выраженное недоразвитие всех отделов верхней челюсти, ложный экзофтальм.
Рото-лице-пальцевой синдром — множественные добавочные уздечки слизистой оболочки рта, порок развития передних двух третей языка, аплазия или синдактилия пальцев рук и ног; сочетается с врожденной расщелиной губы или неба. Тип наследования аутосомно-рецессивный.
Краниосиностоз (краниостеноз, от греч. cranio — череп и synostosis — сращение костей) — раннее закрытие черепных швов, что способствует ограниченному объему черепа, его деформации и внутричерепной гипертензии. Заболевание встречается у одного новорожденного на 2000, чаще наблюдается у мальчиков. (Л.О. Бадалян, 1984).
Дизостоз — (dysostosis; греч. dys- + osteon кость + osis) :чаще всего — аномалии развития костей черепа в сочетании с другими симптомами, однако встречаются множественные и генерализованные поражения костей скелета.
Основные разновидности : черепно-ключичный, черепно-лицевой, челюстно-лицевой и челюстно-черeпной.
Черепно-ключичный дизостоз
Характеризуется гипоплазией покровных костей черепа в сочетании с полным или частичным недоразвитием одной или обеих ключиц.
Симптомы:
незаращение или позднее заращение черепных швов и родничков, брахицефалия (см.) с преобладанием расширения свода черепа в латеральных направлениях, выдающийся лоб,
гипоплазия лицевых костей, верхняя микрогнатия (недоразвитие верхней челюсти), уплощение средней трети лица,
позднее прорезывание зубов,
отсутствие ключиц или частичное недоразвитие
деформациями позвоночника, костей верхних и нижних конечностей, стоп, тазовых костей.
Тип наследования: рецессивный и доминантный.
Черепно-лицевой дизостоз
Черепно-лицевой Дизостоз (синдром Крузона, синдром Апера) характеризуется недоразвитием костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов (в основном венечного, отделяющего лобную кость от теменных и височных).
Симптомы:
изменение формы мозгового отдела черепа («башенный» чепеп),
ложный экзофтальм, косоглазием, нистагм, расстройство зрения,
недоразвитие верхней челюсти (верхняя микрогнатия), уплощение средней зоны лица,
выдающийся нос.
Тип наследования: аутосомно-доминантный
Синдром Апера, симптомы:
карликовый рост,
башенная форма черепа,
расширенная переносица,
расщелина твердого нёба,
синдактилии на руках и ногах.
Челюстно-лицевой дизостоз
Челюстно-лицевой Дизостоз — синдром Берри—Франческетти, синдром Франческетти—Цвалена.
Симптомы:
нижняя микрогнатия, «птичий» профиль лица,
гипоплазия скуловых костей,
макростомия,
антимонголойдный разрез глаз,
слепые фистулы от углов рта к ушам,
языковидное оволосение щек,
нарушения развития зубов,
деформация ушных раковин, иногда среднего и внутреннего уха.
в противоположность синдромам Крузона и Апера — сильное развитие лобных пазух,
деформация грудной клетки и позвоночника.
Тип наследования: аутосомно-доминантный.
Челюстно-черепной Дизостоз
Челюстно-черепной Дизостоз: синдром Петерс — Хевельса.
Симптомы:
верхняя микрогнатия, укорочение прееднего отдела основания черепа,
прогения, открытый прикус.
Другие формы: синдромы Гегенхара, Робена, Франсуа
Тип наследования: аутосомно-доминантный.
Эктодермальная дисплазия
Эктодермальные дисплазии — врождённые дефекты структур эктодермального генеза (в т.ч. кожи и её придатков) — наблюдают в виде нескольких самостоятельных форм и при ряде заболеваний, различающихся по клинической картине.
Основные формы
• Крпста-Сименса-Турена синдром (*305100, Xql3.1, дефект гена EDA, К рецессивное): врождённое сочетание отсутствия потовых желез, частичного или полного отсутствия зубов, гипотрихоза, деформации костей носа, хейлита и синеватой пигментации кожи. У женщин-носительниц при йодной пробе характерно распределение потовых желез в форме спиралей или V-образно, часто более выраженное на одной стороне тела. Синонимы: эктодермальная дисплазия гипо-гидротическая, Крйста-Сйменса синдром, дисплазия эктодермальная ангидротическая, Сйменса синдром, кератоз идиопатический многоформный.
• Дисплазия эктодермальная гидротическая (*129500, 13qll-q12.1, дефект гена HED, R): аномалия развития эктодермы, проявляющаяся дисплазией эпидермиса и придатков кожи -дистрофия зубов, рахит, хейлит, конъюнктивит, врождённая дистрофия ногтей и волос (утолщение ногтей, урежение или отсутствие волос на голове), часто сопровождается ке-ратодермией ладоней и стоп, гиперпигментацией кожи, косоглазием и умственной отсталостью; потоотделение не нарушено. Синонимы: синдром Клустона, Франческётти эктодермальная дисплазия. Клустона гидротическая эктодермальная дисплазия. Примечание. Возможна ассоциация синдрома Клустона с глухотой вследствие протяжённой делеции, захватывающей области гена HEDи гена коннексина (121011, 13ql2). Редкие формы. Различают около 20 форм эктодермальных дисп-лазий (с нарушенным или нормальным потоотделением), например:
• Синдром Кристиансона-Фурй (601375, R) отличается от синдрома Клустона (129500) отсутствием поражения кожи
• Синдром Базана(* 129200,R) — эктодермальная дисплазия без дерматогли-фических узоров ладоней, с изменениями ногтей и четырёхпальце-вой ладонной складкой
• Дисплазия эктодермальная гипогидроти-ческая с гипотиреозом и агенезией мозолистого тела
Источник
Ухо-небно-пальцевой синдром. Ото-палато-дигитальный или OPD-синдромИмеются многочисленные наследственные заболевания, при которых наблюдаются глухота и аномалии костно-мышечной системы. Болезни костной системы распределяются от костных аномалий, охватывающих только несколько костей, таких, как челюстно-лицевой дизостоз и рото-лице-пальцевой синдром II, до генерализованных поражений костной системы, таких, как болезнь Педжета, краниометафизарная дисплазия и склеростеоз. Некоторые из этих синдромов очень редки и встречаются только в одной единственной пораженной семье, например, такие, как глухота и дисгенезия большой берцовой кости. При некоторых синдромах наблюдается проводящая глухота, при других — нейросенсорная или смешанная. OPD-синдром характеризуется проводящей глухотой, задержкой роста, расщеплением неба, лицом «боксера» и общей костной дисплазией. Несколько групп ученых из университета в Миннесоте (Dudding, Gorlin, Langer, Langer, Buran, Duvall) изучили клиническую и рентгенологический) картину синдрома у 3 больных сибсов. Ранее одно наблюдение было описано Taybi. Подробное рентгенологическое исследование большой семьи было проведено Poznanski с соавт.. Клинические данные. Данные осмотра. При рождении ребенок имел нормальную массу и длину, но на протяжении детского возраста его физическое развитие было более замедленным, чем у здоровых детей, масса и рост его были на 10% ниже физиологической нормы. Лицо характеризовалось нависающими бровями с большими надглазничными утолщениями, уплощенной средней частью лица, выступающим затылком, косым антимонголоидным разрезом глаз, гипертелоризмом и широким плоским переносьем. Общий вид больного напоминал боксера. Небо обычно было расщеплено. Могут наблюдаться подвывихи головок лучевой и бедренной костей. Концевые фаланги пальцев широкие, V пальцы короткие с клинодакти-лией; I пальцы на руках были похожи на обрубки, а пальцы на ногах были широкие и кривые, похожие на сучки дерева. Нередко между пальцами имеется перепонка. Орган слуха. Каждый больной считал себя глухим с детского возраста. Аудиометрнческое исследование 3 сибсов показало двустороннюю проводящую глухоту на уровне от 30 до 90 дБ (Buran, Duvall). Двум сибсам была произведена односторонняя тимпанотомия. В каждом случае были обнаружены утолщенные слуховые косточки. У одного сибса был утолщен длинный отросток наковальни и в результате этого сустав между наковальней и стременем был несостоятельным. Головка стремени была расширена, а передняя ножка не достигала основания. У другого сибса ни одна из ножек стремени не достигала основания.
Лабораторные данные. Рентгенограммы. Резко выражены надглазничные бугры. Лобная и затылочная кости утолщены и придают черепу вид гриба. Основание передней черепной ямки также утолщено. Кости лица мелкие, также, как и пазухи верхней челюсти и основной кости. Угол между носовой полостью, турецким седлом и основанием черепа значительно редуцирован. Часто отмечается вывих кзади головки лучевой кости. Па руках видна клинодактилия V пальцев вследствие укорочения средних фаланг со стороны лучевой кости. Дистальные фаланги I—IV пальцев короткие и широкие. Другие аномалии включают добавочный центр окостенения во второй пястной кости и малую многоугольную кость в виде капли слезы. У жепщин-гетерозигот может наблюдаться синостоз между большой многоугольной и ладьевидной костями. Аномалии ног включают боковое искривление бедренных костей и нижней половины большой берцовой кости. Фаланги пальцев на ногах короткие, II и III плюсневые кости в результате слияния с клиновидными костями имеют форму весла; V пястная кость может выступать и содержать дополнительный центр окостенения (Langer, 1967; Gall et al.). Наследственность. Синдром наследуется по Х-сцепленному рецессивному типу. У женщин-гетерозигот наблюдаются выдающиеся боковые части надбровных дуг и малые скелетные аномалии (Gorlin et al., Poznanski et al.). Диагноз. У больных с синдромом Ларсена отмечаются некоторое сходство лица, расщепление неба и вывихи суставов. Однако этот синдром можно отдифференцировать на основании рентгенологических данных (множественные кости запястья, околопяточная кость и т. д.), а также па основании аутосомно-рецессивного или доминантного типа наследования. Лечение. Расщепление неба может быть восстановлено хирургическим путем. Глухоту можно лечить применением слуховых аппаратов или тимпапотомией с введением протезов вместо аномальных слуховых косточек. Выводы. Характеристика этого синдрома включает: 1) Х-сцепленное рецессивное наследование; 2) «лицо боксера», включающее широкий корень носа, гипертелоризм, выступающие лобные и теменные бугры, а также маленькую нижнюю челюсть; 3) расщепление неба; 4) отставание в росте; 5) аномалии рук и ног, включающие широкое расстояние между I и II пальцами и укорочение I пальцев; 6) значительно варьирующие скелетные аномалии; 7) легкую умственную отсталость; 8) умеренную проводящую глухоту. — Также рекомендуем «Синдром Мора. Рото-лице-пальцевой или OFD II синдром» Оглавление темы «Наследственная глухота»:
|
Источник