Родился ребенок с синдромом франческетти как быть

Синдром Франческетти или Тричера Коллинза – врождённое заболевание, которое встречается крайне редко. Согласно статистике, эта патология наблюдается у одного из пятидесяти тысяч новорождённых. Стоит отметить, что существуют несколько стадий развития болезни. Если говорить о начальной, то здесь ребёнку практически ничего не угрожает. У него есть некоторые дефекты внешне, но это никак не отображается на умственном развитии малыша.
Если заболевание протекает без осложнений, то дети с таким синдромом могут вести активный образ жизни. Однако не всё так радужно. В случае тяжёлой стадии развития болезни ребёнок очень сильно страдает, у него практически отсутствует лицо. Это полностью исключает новорождённого из социума. В данной статье мы рассмотрим причины появления синдрома Франческетти, симптомы и методы лечения.
Что это такое?
Как уже было отмечено, синдром Коллинза является исключительно генетическим заболеванием. Его невозможно приобрести в течение жизни, он проявляется с рождения. Данная болезнь – один из подвидов дизостоза, который, в свою очередь, означает нарушение или неправильное развитие костных тканей. Если говорить о синдроме Франческетти, то здесь происходит деформация костей черепа, что не может не оказывать влияние на физическое и моральное состояние человека.

У новорождённого сразу заложены клинические проявления, от количества которых зависит тяжесть развития заболевания. Стоит отметить, что болезнь характеризуется нарушением в строении черепа, которое отображается на лице человека. Чаще всего страдают от синдрома дети, у родителей которых наблюдаются спонтанные мутации генов.
Причины появления болезни
Синдром является врождённым заболеванием, что исключает влияние каких-либо внешних и внутренних факторов на его развитие. Необходимо отметить, что недуг изначально заложен в аминокислотный код ребёнка, причём ещё до его появления на свет. Учёные сумели доказать появление разного рода мутаций генов в пятой хромосоме. Это не случайность, ведь именно она отвечает в организме человека за качество материала для скелета. Кроме того, говоря о строении хромосомы, можно отметить её размер: сама длинная структура в геноме.

Сами по себе мутации происходят на основе нарушения во внутриклеточном синтезе белка. Затем у человека наблюдают гаплонедостаточность. Другими словами, в организме просто не хватает белка для формирования лицевой части черепа. Основной причиной появления синдрома Франческетти является наследственность генов. Однако заболевание может развиться из-за новых мутаций, которые вызываются некоторыми факторами:
- радиоактивное излучение;
- токсоплазмоз;
- приём психотропных медикаментов.
Признаки синдрома
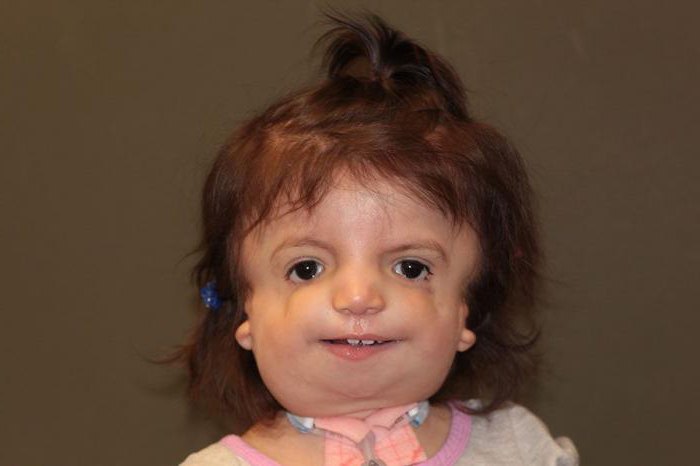
Так как данная болезнь характеризуется тем, что поражает ещё не родившийся плод, то первые симптомы можно обнаружить сразу после появления на свет. Более того, практически все признаки недуга проявляются сразу. Даже несведущий человек в медицине при мимолётном взгляде на больного может выявить определённые симптомы. Главным из них является нарушение формы глаз. Видно сразу человека с синдромом Франческетти — фото, представленные в статье, это наглядно демонстрируют.

Кроме вышеуказанного признака, есть ещё несколько, которые характеризуют данное заболевание. Стоит отметить, что все симптомы довольно ярко выражены, и их сложно не заметить. Итак, среди них наиболее распространёнными являются:
- сбой в строении скул и нижней челюсти;
- проблемы со слухом;
- нарушение в структуре подбородка;
- отсутствие ушных раковин;
- неправильный прикус.
Уровни развития недуга
В начале статьи было указано, что существует несколько стадий болезни, о которых пришло время поговорить подробнее. Синдром Франческетти (Коллинза) имеет три уровня развития. Первый и наиболее безопасный характеризуется небольшой гипоплазией костей лицевой части. Однако даже в этом случае человек меняется внешне, и самые внимательные могут заподозрить наличие заболевания.
Когда наступает вторая стадия, то здесь уже добавляются проблемы со слухом, неправильная форма глазных щелей, деформация нижней челюсти. В этом случае недуг становится более явным, и человек с синдромом внешне значительно отличается от обычного индивида.
Что касается последней, наиболее тяжёлой стадии, то тут имеет место полное отсутствие лица. На самом деле это очень страшно, ведь пациент просто автоматически исключается из общества, и с очень низкой долей вероятности что-то изменится. Кроме того, эти люди с годами страдают сильнее, ведь патология только усугубляется. Могут появиться признаки более тяжёлых болезней. Синдром Франческетти – сам по себе довольно опасный недуг, а с годами могут приобретаться заболевания и похуже.
Наиболее серьёзные осложнения
Помимо того что синдром и так ограничивает многие возможности человека, есть ещё и особые случаи, которые оказывают сильнейшее влияние на здоровье организма. Одним из наиболее тяжёлых осложнений принято считать деформацию ротового аппарата. Это связано с проблемами в строении зубов, что приводит к тому, что больной не может самостоятельно кушать.

Также могут возникнуть дополнительные сложности с дыханием, так как при синдроме Франческетти язык увеличивается в размерах и зарастают носовые проходы. Безусловно, эти факторы оказывают негативное влияние на дыхательную систему человека.
Диагностика
Стоит отметить, что выявление данного недуга происходит во время беременности. У сформировавшегося плода врачи диагностируют наличие синдрома Франческетти. Рекомендуется проводить исследование лицевой части с помощью биопсии. Однако этот способ не является безопасным, и поэтому врачи часто используют для этих целей УЗИ. Также необходимо у всех членов семьи взять анализы крови. После этого беременной женщине следует пройти фетоскопию. Заключительным этапом диагностики является взятие крови из плацентарных сосудов.
Причины синдрома Франческетти кроются в мутации генов, поэтому в зависимости от выраженности болезни врачи принимают решение о целесообразности дальнейших исследований. Если заболевание характеризуется экспрессивностью, то здесь сомнений быть не может. Однако в случае обнаружения некоторых отдельных симптомов специалисты обязаны продолжить наблюдение. Список мероприятий для выявления синдрома составляется врачом индивидуально, и в большинстве случаев это приносит свои плоды. Данное заболевание является довольно тяжёлым, и поэтому очень важно диагностировать его заранее, чтобы оставался шанс на исправление положения вещей.
Лечение синдрома Франческетти
К сожалению, необходимо констатировать тот факт, что в настоящее время медицина не готова предложить терапевтические методы лечения данной проблемы, их попросту не существует. Вся терапия направлена исключительно на паллиативную помощь. Человек, который отличается от всех остальных, сильно желает быть кому-то нужным. Поэтому в этом случае ценнее простой человеческой заботы трудно что-либо найти.

Если наблюдаются тяжёлые стадии заболевания, то следует делать операцию. Если у человека при этом есть проблемы со слухом, врачи рекомендуют использовать слуховой аппарат. Конечно, от моральной поддержки зависит очень многое. Сможет ли пациент поверить в себя? Сможет ли быть сильнее всех проблем? Довольно трудные вопросы. Это говорит о том, что психологическая помощь не может быть лишней никогда. В любой ситуации при возможности необходимо поддержать добрым словом. Вам это ничего не стоит, а больному это может открыть второе дыхание.
Прогноз
Стоит отметить, что в большинстве случаев люди с диагнозом синдром Франческетти проживают полноценную жизнь, находят супругов, рожают детей. Однако, что касается общества, то тут возникают определённые сложности. Социально адаптироваться таким людям нереально трудно, что, безусловно, негативно отражается на их здоровье.

Но если у человека не самая тяжёлая стадия болезни и он морально справился с давлением общественности, то шанс на полноценную жизнь возрастает. Главное – преодолеть проблему психологически и продолжать делать своё дело.
Заключение
Синдром Коллинза встречается довольно редко, однако является серьёзным заболеванием. Причиной данной болезни является наследственность, спонтанная мутация генов. Врачи во время обследований беременной женщины начинают проверку на наличие деформации лицевой части плода. Если исследование показало, что болезнь существует, специалист обязан составить курс терапии, который максимально облегчит жизнь человеку.
Источник
Мандибуло-фациальный дизостоз – это поражение структур, исходящих из первой жаберной дуги, наследуемый по аутосомно-доминантному типу с высокой (до 90%) пенетрантностью и различной экспрессивностью переменной, даже среди больных одной семьи. Может наблюдаться в двух или даже трех генерациях [1].
Этот синдром вызывается мутациями в TCOF1 гене (5q32-q33.1) или в POLR1C гене (6p21.1) и POLR1D гене (13q12.2), кодирующих РНК-полимеразы I и III подразделений. Он обусловлен дисплазией эмбрионального элемента первой жаберной дуги неизвестного происхождения [2]. Описывается, как врожденное нарушение развития костей черепа (чаще височной кости) и лица, характеризуется двусторонней симметричной ото-нижнечелюстной дисплазией без аномалий развития конечностей, он также связан с некоторыми дефектами головы и шеи [3].
Синдом Франческетти — редкое заболевание, встречающееся в 1 случае на 50 000 детей, рожденных живыми.
Дифференциальная диагностика проводится с синдромом глазо-ушно-позвоночной дисплазии Гольденхаара, при которой имеются эпибульбарные дермоиды, часто связанные с колобомой верхнего века, ушные отростки и предушные слюнные фистулы, нарушение строения наружного слухового прохода и тугоухость, шейный синостоз, увеличение количества грудных или поясничных позвонков, и другие аномалии позвоночника. Часто наблюдается высокое расщепленное небо, раздвоенный язык и аномалии зубов [4, 5].
Несмотря на наличие разнообразных аномалий, прогноз при синдроме Франческетти благоприятный. Этиологического лечения не существует, по возможности проводят пластические операции. Гибель пациентов чаще наступает от интеркуррентных заболеваний.
Под нашим наблюдением находилась новорожденная доношенная девочка Мария Г., родившаяся от 6 беременности, на фоне: отеков, вызванных беременностью, хронической внутриутробной гипоксии плода, фетоплацентарной недостаточности, вегето-сосудистой дистонии по гипертоническому типу, хронического гастрита. В 20 недель мать проходила лечение по поводу острого токсоплазмоза, отмечалось многоводие, в 34 недели у ребенка выявлен порок мочевыводящей системы (МВС). Роды 3 срочные в головном предлежании. Масса ребенка при рождении 2800 граммов, рост — 50 см., с оценкой по шкале Апгар 4, 5, 5 баллов.
Из родильного зала ребенок поступил в ОРИТН в тяжелом состоянии за счет явлений дыхательной недостаточности 3 степени, неврологической симптоматики, задержки внутриутробного развития, кожного геморрагического синдрома. С рождения до 20 суток жизни проводилась респираторная терапия, затем, в течение 7 дней дыхание через воздуховод, в последующем — спонтанное, лишь при нарастании дыхательной недостаточности периодически нуждалась в постановке воздуховода. Проводилось зондовое кормление.
Обращали на себя внимание множественные пороки развития: выраженная микрогения, гипоплазия скуловых костей, большой «клювовидный нос», короткий фильтр, глазной гипотеларизм, микрофтальмия, антимонголоидный разрез глаз, «птичье лицо», деформация ушных раковин (низко расположенные, большие уши), гипоплазия ногтевых пластинок (на кистях и стопах), широкая грудная клетка, сосковый гипертеларизм, выраженная бледность кожных покровов. В связи с выявлением пороков развития и более 5 стигм дисэмбриогенеза, ребенок консультирован генетиком. На основании фенотипических данных обследования методом синдромологического анализа был заподозрен синдром Франческетти: аутосомно-доминантное заболевание.
Пациентке было проведено кариотипирование, выявлен нормальный женский кариотип (46 хх).
Ребенок находился на зондовом питании, на третьи сутки жизни девочка была проконсультирована детским хирургом по поводу непроходимости носовых ходов. Поставлен диагноз: ВПР. Атрезия хоан.
Кроме того, дополнительно потребовался осмотр отоларинголога, проведена попытка зондирования носовых ходов зондом №4. Диагноз: Атрезия хоан слева, сужение справа. Было рекомендовано хирургическое лечение в плановом порядке.
Отоакустическая эмиссия у ребенка не была зарегистрирована (заподозрена тугоухость).
На 23 сутки жизни, после стабилизации состояния, девочка была переведена в отделение патологии новорожденных и недоношенных детей для дальнейшего обследования и лечения.
По данным нейросонографии выявлена гипоплазия мозолистого тела, расширение межполушарной щели и субарахноидального пространства, дилатация 3 желудочка мозга.
После чего, девочка была осмотрена неврологом и выставлен диагноз: ВПР головного мозга: Микроцефалия? Гипоплазия мозолистого тела. Последствия гипоксического поражения нервной системы, синдром двигательных нарушений. Задержка психомоторного развития, ранний восстановительный период.
По результатам ДЭХО-КГ выявлена коарктация аорты, функционирующее овальное окно до 0,47 см, открытый артериальный проток — 0,4 см. Диагноз подтвержден кардиохирургом, было рекомендовано оперативное лечение в более позднем периоде.
После осмотра окулистом, у девочки выявлена микрофтальмия, а также атрофия дисков зрительных нервов обоих глаз.
По данным УЗИ брюшной полости обнаружена тазовая дистопия и гипоплазия левой почки. С четвертой недели жизни появились изменения в моче — лейкоцитурия до 25 в поле зрения, в динамике уровень лейкоцитов нарос до 40, появилась протеинурия. В посеве мочи на стерильность выявлен умеренный рост E. Coli, была назначена антибактериальная терапия. Девочка осмотрена детским урологом и, учитывая наличие врожденного порока развития МВС, заподозрено наличие пузырно-мочеточникового рефлюкса в гипоплазированную почку. Было рекомендовано: УЗИ до и после микции, продолжить проведение антибактериальной терапии, поставить постоянный катетер, контроль ОАМ 1 раз в 2 недели, а также наблюдение и обследование ребенка урологом в плановом порядке, после коррекции пороков развития.
Перед выпиской из отделения, ребенок повторно осмотрен генетиком и, с учетом выявленных пороков развития (атрезия хоан слева и сужение справа, сформировавшаяся микроцефалия, гипоплазия мозолистого тела, тазовая дистопия и гипоплазия левой почки, ВПС – коарктация аорты, ОАП, а также множественные стигмы дисэмбриогенеза), синдром Франческетти был подтвержден.
За время нахожнения в стационаре, девочке проводилось симптоматическое лечение, направленное на поддержание витальных функций и усиление адаптационных возможностей организма, антибактериальная, ноотропная, инфузионная терапия и парентеральное питание.
После проведенного лечения и объяснения маме особенностей ухода за ребенком, необходимости хирургической лечения по восстановлению проходимости носовых ходов, коррекции ВПС, необходимости динамического наблюдения узких специалистов, ребенок переведен ОДКБ.
В заключение следует сказать о том, что настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку крайне редко встречается в повседневной практике. Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое нами клиническое наблюдение, представляет большие сложности. По нашему мнению, в подобных ситуациях оправдана постановка синдромологического диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования, с последующей хирургической коррекцией нарушенных функций.
Источник
Синдром Франческетти является аутосомно-доминантным расстройством черепно-лицевого развития с переменной экспрессией. Широко известен как синдром Тришера Коллинза (TCS).
Назван в честь английского офтальмолога Э. Т. Коллинза, который в 1900 году описал основные компоненты этого состояния. Расстройство влияет на оба пола одинаково.
В Англии и на американском континенте эта аномалия описывается как «синдром Тришера Коллинза», на европейском континенте – «Мандибуло-лицевой дизостос» или «синдром Франческетти»
Другие названия:
- Синдром Франческетти-Звален-Клейн;
- Мандибулофациальный дистоз (MFD1);
- Синдром Тришера Коллинза-Франческетти;
- Зигоуроандибулярная дисплазия.

Характеристики
Синдром Франческетти – серьезное врожденное расстройство черепно-лицевого развития, характеризующееся многочисленными аномалиями, которые ограничены головой и лицом. Гипоплазия костей лица, особенно челюстной и скуловой системы, является общей чертой.
Эта болезнь почти полностью отсекает ребенка от общества, делая его жизнь еще более тяжелой и болезненной.
Синдром Франческетти – это генетическое заболевание, вызванное мутацией генов, помогающих правильному развитию костей и лицевых тканей. Этот белок важен для развития лица как в утробе матери, так и после рождения. Мутация приводит к уменьшению его количества, изменяет клетки формирующие черты лица.
Нарушения в развитии костной ткани, вызывают сильную асимметрию, из-за чего слуховой и зрительный аппарат приобретают постоянные структурные патологии.
Распространение
Редкое генетическое заболевание, затрагивает приблизительно 1 из 50 000 человек.
Причины
Развитие заболевания связано с нарушениями внутриутробного развития на 6-7 неделе.
Расстройство возникает из-за мутаций гена TCOF1 или POLR1C, POLR1D. Это аутосомно-доминантное состояние, что означает, что одна копия измененного гена в каждой клетке достаточна, чтобы вызвать расстройство.
Около 60 процентов случаев являются результатом новых мутаций и встречаются у людей, не имеющих истории семейного расстройства. Чаще, человек наследует измененный ген от пострадавшего родителя.
Когда синдром Франческетти вызывается мутациями гена POLR1C, состояние имеет аутосомно-рецессивный характер наследования. Такое наследование означает, что обе копии гена в каждой клетке имеют мутации.
Родители индивидуума с аутосомно-рецессивным состоянием имеют одну копию мутированного гена, но обычно не проявляют признаков и симптомов состояния.
Мутации гена TCOF1 являются наиболее распространенной причиной расстройства, составляя от 81 до 93 процентов всех случаев.
POLR1C и POLR1D вызывают дополнительные 2 процента случаев.
Белки, полученные из генов TCOF1, POLR1C и POLR1D, играют важную роль в раннем развитии костей и других тканей лица. Они участвуют в получении молекулы, называемой рибосомальной РНК (рРНК). Рибосомальная РНК помогает собрать белковые строительные блоки (аминокислоты) в новые белки, которые необходимы для нормального функционирования и выживания клеток.
Мутации TCOF1, POLR1C, POLR1D уменьшают продукцию рРНК. Исследователи предполагают, что уменьшение количества рРНК может спровоцировать саморазрушение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица.
Аномальная гибель клеток приводит к специфическим проблемам развития лица, обнаруженным при синдроме Франческетти.

Признаки
Признаки этого расстройства сильно различаются: от почти незаметного до тяжелого. Симптомы синдрома Франческетти часто обнаруживаются на УЗИ во время беременности. Чаще проявляется деформированное и недоразвитое лицо, подбородок.
Нижние веки поражены, скулы, челюсть, уши могут отсутствовать или иметь дефекты. У детей возникают проблемы с дыханием и слухом.

Симптомы таковы:
Челюсти и зубы
- Расщепление неба (неправильная крыша рта);
- Неполное открытие рта;
- Зубные промежутки;
- Маленькая челюсть;
- Углы рта наклонены вниз.
Лицо
- Отсутствует кость бровей;
- Отсутствующие или маленькие скулы;
- Нисходящие наклонные глаза;
- Нижнее веко опускающееся;
- Расширение области рта.
Уши
- Характеризуется отсутствующими, маленькими или необычно сформированными ушами;
- Отсутствие слухового прохода;
- Уши расположены слишком близко к области шеи.
Потеря слуха вызвана дефектами трех небольших костей в среднем ухе, которые передают звук, или из-за недостаточного развития слухового прохода.

Общие симптомы
- Проблемы с глотанием;
- Нарушения зрения. Часто имеют глаза, которые наклонены вниз, редкие ресницы, выемка на нижних веках, называемая coloboma;
- Потеря слуха;
- Затрудненное дыхание;
- Проблемы с речью;
- Деформации рук.
Некоторые рождаются с отверстием в крыше рта, называемым волчьей пастью. В тяжелых случаях недоразвитие костей лица ограничивает дыхательные пути пострадавшего ребенка, вызывая потенциально опасные для жизни проблемы с дыханием.
Люди с расстройством обычно имеют нормальный интеллект.

Рекомендации по лечению и коррекции
В настоящее время нет препаратов для лечения синдрома Франческетти. Лечение направлено на конкретные потребности каждого ребенка или взрослого. Необходим многодисциплинарный медицинский подход. Главная проблема – нарушение проходимости дыхательных путей, глотание, зрение, слух.
Новорожденным требуется немедленная трахеостомия, если воздушные пути деформированы. Процедура предполагает размещение трубки непосредственно в области шеи, горла.
Потеря слуха рассматривается как усиление костной проводимости путем размещения имплантов, рекомендуется речевая терапия, образовательные программы.
Во многих случаях необходима черепно-лицевая реконструкция. Хирургия выполняется для восстановления расщепления неба, восстановления челюсти или других костей черепа. Конкретные хирургические процедуры и возраст при проведении операции зависят от тяжести аномалий, общего состояния здоровья, личных предпочтений.
 На фотографии девушка до и после пластической хирургии. Поэтому не отчаивайтесь и не опускайте руки. Всегда возможно найти способ улучшить свой внешний вид.
На фотографии девушка до и после пластической хирургии. Поэтому не отчаивайтесь и не опускайте руки. Всегда возможно найти способ улучшить свой внешний вид.
Костные трансплантаты устанавливаются, чтобы помочь создать скулы. Некоторые из операций откладываются до достижения ребенком 5 лет и старше.
В некоторых случаях невозможно полностью исправить деформации лица, поэтому врачи слегка уменьшают проявление симптомов улучшая качество жизни пациента.
Попытки восстановить слух хирургическим путем (для коррекции структуры слуховых косточек) не показали положительного результата, поэтому рекомендуется использовать слуховые аппараты, выбранные для индивидуальных патологий внутреннего и среднего уха.
Новые методы
Есть несколько возможных методов лечения, которые исследуются. Ученые ищут способы ингибировать протеин p53, который помогает организму убивать нежелательные клетки. У людей с синдромом Франческетти, p53 аномально активируется, что приводит к потере определенных клеток, вызывает симптоматику.
Есть предположение, что ингибирование продуцирования р53 (блокирование его активации) может помочь лечить пораженных людей. Необходимы дополнительные исследования, чтобы определить, эффективен ли этот тип лечения и безопасен ли он.
Исследователи также изучают использование стволовых клеток, обнаруженных в жировой ткани, которые используются при операциях у людей с черепно-лицевыми патологиями.
Ранние исследования показали, что хирургические результаты улучшаются с использованием этих стволовых клеток, помогают стимулировать восстановление роста пораженных участков. Однако подобная терапия все еще экспериментальная и противоречивая.

Прогноз и продолжительность жизни
Дети, рожденные с тяжелыми дефектами, которые влияют на дыхание, умирают вскоре после рождения. Некоторые могут быть спасены при немедленной трахеостомии для возможности дыхания.
Врачи, вскоре после рождения, часто выполняют операцию, которая двигает челюсть в правильное положение, чтобы ребенок смог дышать сам по себе раньше. Это улучшает ожидаемую продолжительность жизни для новорожденных.
Обычно люди с синдромом Франческетти становятся работоспособными взрослыми с нормальным интеллектом. В некоторых случаях прогноз зависит от конкретных симптомов и тяжести заболевания. Очень тяжелые случаи могут вызывать перинатальную смерть из-за патологии дыхательных путей.
Основной проблемой синдрома является социальная адаптация.
Ожидаемая продолжительность жизни для людей с синдромом Франческетти такая же, как и у всех, если все осложнения будут устранены на ранней стадии (дыхательные пути, решение проблем кормления, слуха, зрения). Они имеют нормальный интеллект и могут вести продуктивную жизнь.
По Д-р J Jaranson
Понравилась статья? Поделись с друзьями:
Источник