Робертсоновские транслокации при синдроме дауна

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 11 февраля 2019;
проверки требуют 2 правки.
Робертсоновская транслокация, или центрическое слияние, — хромосомная перестройка, при которой происходит слияние двух акроцентрических хромосом с образованием одной метацентрической или субметацентрической хромосомы. Слияние акроцентриков происходит в околоцентромерных районах, то есть в этой перестройке происходит транслокация целого плеча. Робертсоновские транслокации относятся к межхромосомным перестройкам. Робертсоновские транслокации играют роль в видообразовании, являются частым механизмом эволюции кариотипа, носительство робертсоновских транслокаций может приводить к нарушению фертильности[1].
Данный тип хромосомных мутаций назван по имени американского генетика насекомых Уильяма Робертсона (W. Robertson, 1881—1941), который в 1916 году при сравнении кариотипов близких видов саранчовых впервые предположил существование такого типа хромосомных перестроек[2][3].
В медицинской генетике согласно Международной системе по цитогенетической номенклатуре человека (The International System for Human Cytogenetic Nomenclature — ISCN) для робертсоновских транслокаций используется сокращение der или rob, например, der(13;14)(q10;q10) или rob(13;14)(q10;q10)[4].
Робертсоновские транслокации у людей[править | править код]
Робертсоновские транслокации являются наиболее распространённым видом хромосомных нарушений, встречающимся у людей. Частота носительства робертсоновских транслокаций составляет около 1 случая на тысячу человек[5]. Робертсоновские транслокации у людей являются рекуррентными, то есть регулярно возникающими de novo вследствие структурных особенностей генома человека. Образование робертсоновских транслокаций, как считается, провоцируют повторы сателлитной ДНК III, находящиеся в коротких плечах акроцентриков[6]. Частота возникновения робертсоновских транслокаций de novo является очень высокой и составляет около 4 событий на 10 тысяч гамет на поколение, это на два порядка превышает частоту возникновения мутаций, ведущих к аутосомным доминантным наследственным заболеваниям. Большинство робертсоновских транслокаций возникают во время мейоза в оогенезе[7][8].
У человека хромосомы 13, 14, 15, 21 и 22 являются акроцентрическими, и описаны все возможные комбинации робертсоновских транслокаций с их участием. У этих акроцентриков короткие плечи имеют спутники. В спутничных нитях находятся множественные повторы генов, кодирующих рибосомную РНК. Благодаря множественности повторов рДНК (англ.)русск., потеря в перестройке коротких плеч не приводит к значимому геномному дисбалансу, поэтому робертсоновские транслокации совместимы с нормальным развитием[9]. Участие различных акроцентриков в робертсоновских транслокациях является неравномерным, и наиболее часто встречается носительство робертсоновских транслокаций с участием хромосом 13 и 14 , 14 и 21 (rob(13q14q) и rob(14q21q), соответственно). Эти перестройки составляют около 85 % всех случаев робертсоновских перестроек у людей, при этом около 75 % случаев приходится на транслокацию rob(13q14q)[5].
Носители робертсоновских перестроек могут испытывать проблемы с фертильностью, а их потомство болеть хромосомными болезнями. Однако зафиксирован факт практически бессимптомной передачи транслокации t(13;14) на протяжении 9 поколений одной семьи. Этой семье принадлежит здоровая фертильная женщина с 44 хромосомами, гомозиготная по данной транслокации. Носительство транслокации не сказывалось на фертильности, и в этой семье зафиксирован только один случай рождения ребёнка с трисомией по хромосоме 13[10].
Робертсоновские транслокации у мышей[править | править код]
В нормальном кариотипе домовой мыши (Mus Musculus domesticus) 40 хромосом, все из которых являются акроцентрическими. Цитогенетический анализ мышей из диких популяций, обитающих в некоторых районах Западной Европы (в Швейцарии, Италии, Испании, Германии, на острове Сицилия и др.), выявил популяции мышей с центрическими слияниями[11].
Робертсоновский полиморфизм у обыкновенной бурозубки[править | править код]
Необычный полиморфизм по робертсоновским транслокациям наблюдается у обыкновенной бурозубки (Sorex araneus), широко расселённой по территории Евразии. В отличие от подавляющего большинства видов, у которых кариотип отличается постоянством, число хромосом в различных популяциях бурозубки варьирует от 20 до 33, хотя число хромосомных плеч (фундаментальное число) при этом не меняется и равняется 40[12]. Цитогенетический анализ с использованием дифференциального GTG-окрашивания показал, что кариотип бурозубки состоит из инвариантной и вариабельной частей. Постоянная составляющая кариотипа бурозубки — это 3 пары метацентриков, неизменные в различных популяциях. Вариабельная часть кариотипа бурозубки состоит из 12 пар акроцентрических хромосом, составляющих различные комбинации в центрических слияниях. Группы популяций бурозубки, характеризующиеся одинаковым вариантом кариотипа и населяющих единую территорию, относят к одной хромосомной расе[13]. Всего у бурозубки описано более 70 хромосомных рас[14]. Следует отметить, что разнообразие вариантов кариотипа бурозубки сформировано не только центрическими слияниями, но и реципрокными транслокациями, в которых хромосомы обменивались целыми плечами. Реципрокные полноплечевые транслокации характерны для кариотипов бурозубки, «обогащённых» метацентриками[13].
Робертсоновские транслокации в эволюции кариотипа[править | править код]
Робертсоновские транслокации являются частым механизмом эволюции кариотипа. Например, все 38 аутосом у собаки (Canis familiaris) являются акроцентриками, в то время как у рыжей лисицы (Vulpes vulpes), другого представителя псовых, все 16 пар аутосом являются метацентриками. Считается, что эти два вида имели общего предка примерно 10 млн лет назад. Цитогенетический анализ показал, что происхождение 8-ми метацентриков лисицы можно интерпретировать как продукт центрических слияний акроцентриков предковой формы, кариотип которой, вероятно, был более похож на кариотип собаки[15][16].
Примечания[править | править код]
- ↑ Коряков Д.Е., Жимулев И.Ф. Хромосомы. Структура и функции. — Новосибирск: Из-во СО РАН, 2009. — 258 с. — ISBN 978-5-7692-1045-7.
- ↑ Robertson W. R. B. Chromosome studies I. Taxonomic relationships shown in the chromosomes of Tettigidae: V-shaped chromosomes and their significance in Acrididae, Locustidae, and Gryllidae: Chromosomes and variation // J. Morph. — 1916. — Т. 27. — С. 179-331.
- ↑ МакКонки Э. Геном человека. — М: Техносфера, 2011. — 288 с. — ISBN 978-5-94836-145-1.
- ↑ Баранов В. С., Кузнецова Т. В. Цитогенетика эмбрионального развития человека: научно-практические аспекты / В. С. Баранов, Т. В. Кузнецова. — СПб: Издательство Н-Л, 2007. — 640 с. — ISBN 5-94869-034-2.
- ↑ 1 2 Shaffer L. G., Lupski J. R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. (англ.) // Annual review of genetics. — 2000. — Vol. 34. — P. 297—329. — doi:10.1146/annurev.genet.34.1.297. — PMID 11092830.
- ↑ Jarmuz-Szymczak M., Janiszewska J., Szyfter K., Shaffer L. G. Narrowing the localization of the region breakpoint in most frequent Robertsonian translocations. (англ.) // Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. — 2014. — Vol. 22, no. 4. — P. 517—532. — doi:10.1007/s10577-014-9439-3. — PMID 25179263.
- ↑ Page S. L., Shaffer L. G. Nonhomologous Robertsonian translocations form predominantly during female meiosis. (англ.) // Nature genetics. — 1997. — Vol. 15, no. 3. — P. 231—232. — doi:10.1038/ng0397-231. — PMID 9054929.
- ↑ Bandyopadhyay R., Heller A., Knox-DuBois C., McCaskill C., Berend S. A., Page S. L., Shaffer L. G. Parental origin and timing of de novo Robertsonian translocation formation. (англ.) // American journal of human genetics. — 2002. — Vol. 71, no. 6. — P. 1456—1462. — doi:10.1086/344662. — PMID 12424707.
- ↑ Транслокации хромосом: реципрокные и робертсоновские. Характеристика.. MedUniver.com. Дата обращения 25 февраля 2017.
- ↑ Eklund A1, Simola KO, Ryynänen M., «Translocation t(13;14) in nine generations with a case of translocation homozygosity.», Clinical Genetics, 1988 Feb;33(2):83-6, PMID 3359671
- ↑ Capanna E., Castiglia R. Chromosomes and speciation in Mus musculus domesticus. (англ.) // Cytogenetic and genome research. — 2004. — Vol. 105, no. 2-4. — P. 375—384. — doi:10.1159/000078210. — PMID 15237225.
- ↑ Wójcik J.M., Borodin P.M., Fedyk S., Fredga K., Hausser J., Mishta A., Orlov V.N. The list of the chromosome races of the common shrew Sorex araneus (updated 2002) // Mammalia. — Vol. 67. — P. 169-178. — ISSN 0025-1461. — doi:10.1515/mamm.2003.67.2.169.
- ↑ 1 2 Бородин П. М., Поляков А. В. Хромосомный «портрет» бурозубки на фоне ледников // Природа. — 2001. — № 1. — С. 34-40.
- ↑ Щипанов Н. А. и др. Обыкновенная бурозубка (Sorex araneus) — модельный вид эколого-эволюционных исследований // Зоол. журн.. — 2009. — Т. 88, № 8. — С. 975-989.
- ↑ Yang F., O’Brien P. C., Milne B. S., Graphodatsky A. S., Solanky N., Trifonov V., Rens W., Sargan D., Ferguson-Smith M. A. A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. (англ.) // Genomics. — 1999. — Vol. 62, no. 2. — P. 189—202. — doi:10.1006/geno.1999.5989. — PMID 10610712.
- ↑ Ferguson-Smith M. A., Trifonov V. Mammalian karyotype evolution. (англ.) // Nature reviews. Genetics. — 2007. — Vol. 8, no. 12. — P. 950—962. — doi:10.1038/nrg2199. — PMID 18007651.
Литература[править | править код]
- Баранов В. С., Кузнецова Т. В. Цитогенетика эмбрионального развития человека: научно-практические аспекты / В. С. Баранов, Т. В. Кузнецова. — СПб: Издательство Н-Л, 2007. — 640 с. — ISBN 5-94869-034-2.
- Бородин П. М., Поляков А. В. Хромосомный «портрет» бурозубки на фоне ледников // Природа. — 2001. — № 1. — С. 34-40.
Источник
Генетика синдрома Дауна: кариотипКлинический диагноз синдрома Дауна обычно не представляет никаких трудностей. Тем не менее для подтверждения диагноза и предоставления базы для генетического консультирования необходимо кариотипирование. Хотя различия в конкретных вариантах кариотипа, ответственных за синдром Дауна, обычно имеют небольшое влияние на фенотип пациента, они существенны для определения риска повторения. Трисомия 21 при синдроме Дауна. Примерно у 95% всех пациентов с синдромом Дауна выявляют трисомию хромосомы 21, вызванную мейотическим нерасхождением 21 пары хромосом, как обсуждалось в предыдущей главе. Уже отмечено, что риск иметь ребенка с трисомией 21 увеличивается с возрастом матери, особенно после 30 лет. Мейотическая ошибка, ответственная за трисомию, обычно происходит в ходе материнского мейоза (около 90% случаев), преимущественно в первом делении, но около 10% случаев происходит в отцовском мейозе, обычно во втором делении. Робертсоновская транслокация при синдроме Дауна. Около 4% пациентов с синдромом Дауна имеют 46 хромосом, одна из которых — робертсоновская транслокация между хромосомой 21q и длинным плечом одной из других акроцентрических хромосом (обычно хромосомы 14 или 22). Транслоцированная хромосома заменяет одну из нормальных акроцентрических хромосом, и кариотип пациента с робертсоновской транслокацией между хромосомами 14 и 21 — 46,XX/XY,rob(14;21)(ql0;ql0),+21. Такая хромосома может также быть определена как der(14;21), на практике используют обе номенклатуры. В действительности пациенты с робертсоновской транслокацией, включающей хромосому 21, трисомны по генам, расположенным в длинном плече 21q. В отличие от стандартной трисомии 21, транслокационный синдром Дауна не показывает никакой связи с возрастом матери, но имеет сравнительно высокий риск повторения в семьях, если один из родителей, особенно мать, — носитель транслокации. По этой причине для точного генетического консультирования важно кариотипирование родителей и, возможно, других родственников.
Носители робертсоновской транслокации, включающей хромосомы 14 и 21, имеют только 45 хромосом; одна 14 и одна 21 отсутствуют и заменены транслоцированной хромосомой. Теоретически возможны шесть типов гамет, но три из них не могут привести к жизнеспособному потомству. Три типа гамет жизнеспособные, нормальные, сбалансированные и несбалансированные, имеющие как транслоцированную, так и нормальную хромосому 21. В комбинации с нормальной гаметой это может приводить к зачатию ребенка с транслокационным синдромом Дауна. Теоретически эти три типа гамет производятся в равных количествах, таким образом, теоретический риск ребенка с синдромом Дауна должен быть 1 к 3. Тем не менее расширенные популяционные исследования показали, что несбалансированные хромосомные наборы появляются только у 10-15% потомства матерей и только у нескольких процентов потомства отцов, несущих транслокации, включающие хромосому 21. Транслокация 21q21q при синдроме Дауна. Хромосомная транслокация 21q21q — хромосома, сформированная из двух длинных плеч хромосомы 21; бывает у нескольких процентов пациентов с синдромом Дауна. Считают, что они появляются как изохромосомы, а не робертсоновские транслокации. Большинство таких случаев возникают постзиготически, соответственно, риск повторения низкий. Тем не менее особенно важно убедиться, не является ли родитель носителем (возможно, мозаичным) данной транслокации, поскольку все гаметы носителя такой хромосомы должны также содержать 21q21q хромосому, с двойной дозой генетического материала хромосомы 21, или не иметь хромосомы 21 совсем. Потенциальное потомство, следовательно, неизбежно имеет или синдром Дауна, или нежизнеспособную моносомию 21. Мозаичные носители имеют повышенный риск повторения, таким образом, пренатальная диагностика необходима при всех последующих беременностях. Мозаичный синдром Дауна. Около 2% пациентов с синдромом Дауна — мозаики, обычно с популяциями нормальных клеток и с трисомией 21. Фенотип может быть мягче, чем при типичной трисомии 21. Вообще существует широкая изменчивость в фенотипах мозаичных пациентов, вероятно, отражая различные пропорции трисомных клеток у эмбриона на ранних стадиях развития. Возможно, пациенты с установленным мозаичным синдромом Дауна отражают только клинически более серьезные случаи, поскольку в легких случаях кариотипирование менее вероятно. Частичная трисомия 21 при синдроме Дауна. Очень редко синдром Дауна диагностируют у пациентов, имеющих трисомию только по части длинного плеча хромосомы 21, и еще реже выявляют пациентов с синдромом Дауна без цитогенетически видимой хромосомной аномалии. Такие случаи представляют определенный интерес, поскольку могут указывать, какая область хромосомы 21, вероятно, ответственна за специфические компоненты фенотипа синдрома Дауна и какие области могут утраиваться, не вызывая фенотипических проявлений. Хотя хромосома 21 содержит только несколько сотен генов, попытки согласовывать тройную дозу специфических генов со специфическими аспектами фенотипа синдрома Дауна пока имеют ограниченный успех. Наиболее примечательной стала идентификация области, критической для пороков сердца, наблюдаемых примерно у 40% пациентов с синдромом Дауна. Поиск конкретных генов, существенных для проявления фенотипа синдрома Дауна, среди случайно находящихся рядом с ними в хромосоме 21, — главная задача современных исследований, особенно на мышах в качестве модели. Потенциально перспективное направление — исследование генно-инженерных мышей с дополнительной дозой генов из хромосомы 21 человека (или даже с полной копией хромосомы 21). Такие мыши могут проявлять фенотипические аномалии в поведении, функциях мозга и формировании сердца. — Также рекомендуем «Причины синдрома Дауна. Риск рождения ребенка с трисомией 21″ Оглавление темы «Хромосомные аномалии»:
|

Источник
Весь объем генетического материала заложен всего в 46 парах хромосом. А хромосомы, как известно из биологии, находятся в ядре клетки. Здоровый человек имеет кариотип из 23 пар диплоидных хромосом. То есть 46 ХХ — хромосомный набор женщины, а 46 ХУ — мужской набор хромосом. При разрыве какой-нибудь хромосомы, основной «носительницы» генетического кода, случаются различного рода нарушения.
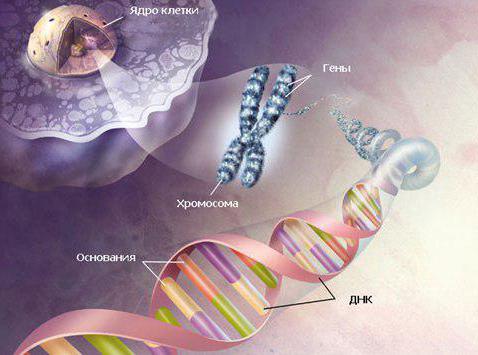
Мутации присущи не только человеку. Небольшие изменения генного материала способствуют разнообразию проявления природы. При так называемой сбалансированной транслокации изменение в хромосомах происходит без потери информации и без лишнего дублирования. Чаще всего это случается при мейозе (делении хромосомы), кроме того, иногда части хромосом дублируются (происходит дупликация), и тогда последствия непредсказуемы. Но мы рассмотрим только робертсоновские транслокации, их особенности и последствия.
Робертсоновские транслокации — что это? Генные проблемы человечества
Вследствие разрыва хромосомы неподалеку от центромеры происходят структурные изменения в генетическом коде человека. Разрыв может быть единичным, а бывает и повторным. Одно плечо хромосомы после разрыва (чаще короткое плечо) теряется. Но попадаются случаи, когда разрыв происходит одновременно в 2 хромосомах, короткие плечи которых меняются местами. Бывает, что подвергаются транслокации только отдельные части плеча. Но такие короткие плечи в хромосомах акроцентрического типа (в которых центромера делит хромосому на более длинное и короткое плечи) никогда не несут жизненно важной информации. К тому же утеря таких элементов не так важна, поскольку этот наследственный материал копируется в других акроцентрических хромосомах.
Но когда отделившиеся короткие плечи срастаются с короткими плечами иного гена, а оставшиеся длинные также спаиваются между собой, то такая транслокация уже не является сбалансированной. Такие «перестановки» генетического материала — это и есть робертсоновские транслокациии.
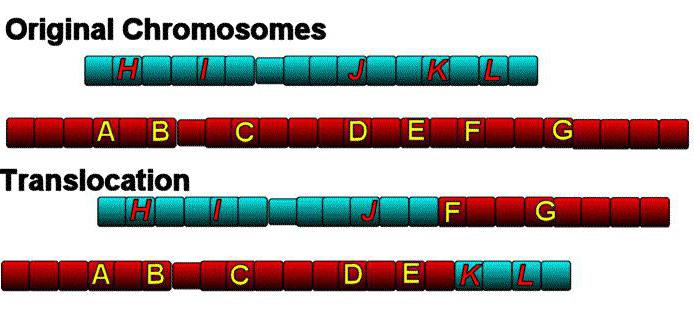
Исследовал и описал такой вид транслокации У. Робертсон в 1916 году. И его именем и была названа аномалия. Робертсоновская транслокация может привести к развитию онкозаболевания, но может и никак не сказаться на внешнем виде и здоровье носителя. Однако ребенок в большинстве случаев, если один из родителей имеет такую транслокацию, рождается с отклонениями.
Насколько часто встречается мутация?
Благодаря усовершенствованию техники и развитию генетики как науки, сегодня можно заранее узнать, есть ли аномалии в кариотипе будущего ребенка. Теперь появилась возможность провести статистику: насколько часто появляются генные аномалии? По современным данным, робертсоновские транслокации встречаются у одного новорожденного из тысячи. Чаще всего диагностируется транслокация 21 хромосомы.

Небольшие хромосомные транслокации абсолютно ничем не угрожают самому носителю. Но когда затрагиваются важные элементы кода, ребенок может родиться мертвым или погибнуть через несколько месяцев, как, к примеру, бывает при синдроме Патау. Но синдром Патау встречается очень редко. Где-то 1 случай на 15 тысяч рождений.
Факторы, способствующие появлению транслокации в хромосомах
В природе существуют спонтанные мутации, то есть ничем не вызванные. Но окружающая среда вносит свои коррективы в развитие генома. Некоторые факторы способствуют учащению мутационных изменений. Эти факторы принято называть мутагенными. Известны следующие факторы:
- воздействие азотистых оснований;
- чуждых ДНК биополимеров;
- прием алкоголя матерью в период беременности;
- влияние вирусов во время беременности.
Наиболее часто происходит транслокация из-за вредного воздействия облучения на организм. Влияет ультрафиолетовое излучение, протонное и рентгеновское излучение, а также гамма-лучи.
Какие хромосомы подвергаются изменениям?
Подвергаются транслокации хромосомы 13, 14, 15 и 21. Самая популярная и опасная транслокация — это робертсоновская транслокация между 14 и 21 хромосомами.
Если в результате мейоза образуется дополнительная хромосома (трисомия) у плода с такой транслокацией, ребенок родится с синдромом Дауна. Такой же прецедент возможен, если произошла робертсоновская транслокация между 15 и 21 хромосомами.
Транслокация хромосом группы D
Робертсоновская транслокация хромосом группы D затрагивает только акроцентрические хромосомы. Хромосомы 13 и 14 участвуют в транслокациях в 74% случаев и их называют несбалансированными транслокациями, которые зачастую опасных последствий для жизни не имеют.
Впрочем, есть одно обстоятельство, которое может сопутствовать подобным аномалиям. Робертсоновская транслокация 13, 14 у мужчин может привести к нарушению фертильности такого носителя-мужчины (хромосомный набор 45 ХУ). Из-за того, что вследствие утери обоих коротких плеч вместо 2 пар хромосом чаще остается только одна, имеющая 2 длинных, гаметы такого мужчины не могут дать жизнеспособного потомства.
Такая же робертсоновская транслокация 13, 14 у женщины также снижает ее возможность родить ребенка. Месячные присутствуют у таких женщин, и все же бывали случаи, когда они рожали здоровых детей. Но статистика все же показывает, что это редкие случаи. В основном их дети нежизнеспособны.
Последствия транслокаций
Мы уже выяснили, что некоторые структурные изменения вполне нормальны и не несут угрозы. Единичная робертсоновская транслокация определяется только благодаря анализам. Но повторная транслокация в наборе хромосом следующего поколения уже опасна.
Робертсоновская транслокация 15 и 21 в сочетании с иными структурными изменениями могут быть даже плачевными. Все последствия отдельных структурных изменений кариотипа опишем более подробно. Напомним, что кариотип — это присущий индивиду набор хромосом в ядре.
Трисомии и транслокации
Кроме транслокаций, генетики выделяют такую аномалию, как трисомия в хромосоме. Трисомия означает, что кариотип плода имеет триплоидный набор одной из хромосом, вместо положенных 2 копий иногда имеет место мозаичная трисомия. То есть триплоидный набор наблюдается не во всех клетках организма.
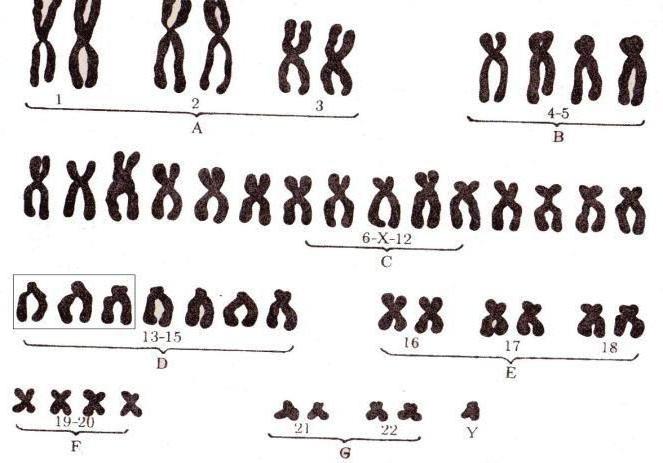
Трисомия в сочетании с робертсоновской транслокацией приводит к очень тяжелым последствиям: таким как синдром Патау, Эдвардса и более распространенный синдром Дауна. В некоторых случаях набор таких аномалий приводит к выкидышу на ранних сроках.
Синдром Дауна. Проявления
Нужно заметить, что транслокации с участием 21 и 22 хромосом более устойчивы. Такие аномалии не приводят к летальным исходам, не являются полулетальными, но просто приводят к отклонению в развитии. Так, трисомия 21 в сочетании с робертсоновской транслокацией в кариотипе при анализе кариотипа плода — это явный «знак» синдрома Дауна, генетического заболевания.
Синдром Дауна характеризуется и физическими и умственными отклонениями. Прогноз жизни у таких людей благоприятен. Несмотря на пороки сердца и некоторые физиологические изменения скелета, их организм функционирует нормально.
Характерные признаки синдрома:
- плоское лицо;
- увеличенный язык;
- много кожи на шее, собирающейся в складки;
- клинодактилия (кривизна пальцев);
- эпикантус;
- порок сердца возможен в 40% случаев.
Люди с таким синдромом медленнее начинают ходить, произносить слова. И также учиться им сложнее, чем иным детям такого же возраста.
Все же они способны на плодотворную работу в обществе и при определенной поддержке и правильной работе с такими детьми в будущем они хорошо социализируются.
Синдром Патау
Синдром встречается реже, чем синдром Дауна, но пороков различного рода у такого ребенка очень много. Практически 80% детей с таким диагнозом погибает в течение 1 года жизни.
В 1960 году изучил эту аномалию и выяснил причины генетического сбоя Клаус Патау, хотя до него в 1657 году описал синдром Т. Бартолини. Риск подобных нарушений увеличивается у тех женщин, которые рожают ребенка после 31 года.

У таких детей многочисленные физические пороки сочетаются с тяжелым нарушением развития психомоторики. Характерны для синдрома:
- микроцефалия;
- аномальные кисти рук, часто образуются лишние пальцы;
- низко посаженные уши неправильной формы;
- заячья губа;
- короткая шея;
- узкие глаза;
- явно «запавшая» переносица;
- пороки почек и сердца;
- расщелина губы или неба;
- при беременности имеется только одна пуповинная артерия.
Небольшому числу выживших младенцев оказывается медицинская помощь. И они способны еще долго жить. Но врожденные аномалии всё-таки сказываются на характере жизни и ее непродолжительности.
Синдром Эдвардса
Трисомия хромосомы 18 на фоне транслокации приводит к синдрому Эдвардса. Этот синдром менее известен. При таком диагнозе ребенок едва доживает до полугода. Закон естественного отбора не позволит развиваться существу с многочисленными отклонениями.
В целом количество различных пороков при синдроме Эдвардса — около 150. Наличествуют пороки развития кровеносных сосудов, сердца, внутренних органов. Всегда присутствует у таких новорожденных гипоплазия мозжечка. Возможны аномалии строения пальцев рук. Очень часто проявляется такая отличительная аномалия, как деформация стопы.
Какие анализы определяют аномалии в период внутриутробного развития?
Для проведения анализа на кариотип плода необходимо получить материал – клетки плода.
Анализов несколько. Осветим, как это все происходит.
1. Биопсия ворсин хориона. Проводится анализ на 10 неделе. Эти ворсины — являются непосредственной частицей плаценты. Эта частица биологического материала все расскажет о будущем плоде.
2. Амниоцентез. С помощью иглы берется несколько клеток плода и амниотическая жидкость. Они берутся чаще всего на 16 неделе беременности, и через несколько недель пара может получить детальные сведения о благополучии ребенка.

На такой анализ направляются матери, у которых риск родить ребенка с отклонениями повышен. Обычно на генетический анализ направляют те пары, у которых:
1) были беспричинные выкидыши;
2) пара долго не могла зачать ребенка;
3) в роду присутствовали связи близкородственного характера.
Такие молодые люди, возможно, имеют робертсоновские транслокации какой-то хромосомы. И поэтому они должны заранее сделать анализ на свой кариотип, чтобы знать, какие есть шансы выносить и родить здорового ребенка.
Источник