Ретикулоцитоз код мкб 10

- D70 Агранулоцитоз. Агранулоцитарная ангина. Детский генетический агранулоцитоз. Болезнь Костманна. Нейтропения: БДУ, врожденная, циклическая, медикаментозная, периодическая, селезеночная (первичная), токсическая. Нейтропеническая спленомегалия. При необходимости идентифицировать лекарственное средство, вызвавшее нейтропению, используют дополнительный код внешних причин (класс ХХ).
- Исключена: преходящая неонатальная нейтропения (P61.5)
- D71 Функциональные нарушения полиморфно-ядерных нейтросфилов. Дефект рецепторного комплекса клеточной мембраны. Хронический (детский) гранулематоз. Врожденный дисфагоцитоз. Прогрессирующий септический гранулематоз.
- D72 Другие нарушения белых кровяных клеток.
- Исключены: базофилия (D75.8), иммунные нарушения (D80 — D89), нейтропения (D70), предлейкоз (синдром) (D46.9)
- D72.0 Генетические аномалии лейкоцитов. Аномалия (грануляции) (гранулоцита) или синдром: Альдера, Мая — Хегглина, Пельгера — Хюэта. Наследственная лейкоцитарная: гиперсегментация, гипосегментация, лейкомеланопатия.
- Исключен: синдром Чедиака — Хигаши (Chediak — Higashi syndrome) (- Стейнбринка) (E70.3)
- D72.1 Эозинофилия. Эозинофилия: аллергическая, наследственная.
- D72.8 Другие уточнённые нарушения белых кровяных клеток. Лейкемоидная реакция: лимфоцитарная, моноцитарная, миелоцитарная. Лейкоцитоз. Лимфоцитоз (симптоматический). Лимфопения. Моноцитоз (симптоматический). Плазмацитоз.
- D72.9 Нарушение белых кровяных клеток неуточненное
- D73 Болезни селезенки
- D73.0 Гипоспленизм. Аспления послеоперационная. Атрофия селезенки.
- Исключена: аспления (врожденная) (Q89.0)
- D73.1 Гиперспленизм.
- Исключены спленомегалия: БДУ (R16.1), врожденная (Q89.0).
- D73.2 Хроническая застойная спленомегалия
- D73.3 Абсцесс селезенки
- D73.4 Киста селезенки
- D73.5 Инфаркт селезенки. Разрыв селезенки нетравматический. Перекручивание селезенки.
- Исключен: травматический разрыв селезенки (S36.0)
- D73.8 Другие болезни селезенки. Фиброз селезенки БДУ. Периспленит. Спленит БДУ.
- D73.9 Болезнь селезенки неуточнённая
- D74 Метгемоглобинемия
- D74.0 Врожденная метгемоглобиемия. Врожденная недостаточность NADH-метгемоглобинредуктазы. Гемоглобиноз М (болезнь НЬ-М). Meтгeмoглобинемия наследственная
- D74.8 Другие метгемоглобинемии. Приобретённая метгемоглобинемия (с сульфгемоглобинемией). Токсическая метгемоглобинемия
- D74.9 Метгемоглобинемия неуточнённая
- D75 Другие болезни крови и кроветворных органов.
- Исключены: увеличение лимфатических узлов (R59.-), гипергаммаглобулинемия БДУ (D89.2), лимфаденит; БДУ (I88.9), острый (L04.-), хронический (I88.1), брыжеечный (острый) (хронический) (I88.0)
- D75.0 Семейный эритроцитоз. Полицитемия: доброкачественная, семейная.
- Исключен: наследственный овалоцитоз (D58.1)
- D75.1 Вторичная полицитемия. Полицитемия: приобретённая, связанная с: эритропоэтинами, снижением объёма плазмы, высотой, стрессом, эмоциональная, гипоксемическая, нефрогенная, относительная.
- Исключены полицитемия: новорождённого (P61.1), истинная (D45)
- D75.2 Эссенциальный тромбоцитоз. Исключена: эссенциальная (геморрагическая) тромбоцитемия (D47.3)
- D75.8 Другие уточнённые болезни крови и кроветворных органов. Базофилия
- D75.9 Болезнь крови и кроветворных органов неуточнённая
- D76 Отдельные болезни, протекающие с вовлечением лимфоретикулярной ткани и ретикулогистиоцитарной системы.
- Исключены: болезнь Леттерера — Сиве (C96.0), злокачественный гистиоцитоз (C96.1), ретикулоэндотелиоз или ретикулез: гистиоцитарный медуллярный (C96.1), лейкемический (C91.4), липомеланотический (I89.8), злокачественный (C85.7), нелипидный (C96.0)
- D76.0 Гистиоцитоз из клеток Лангерганса, не классифицированный в других рубриках. Эозинофильная гранулема. Болезнь Хенда — Шюллера — Крисчена (Hand — Schuller — Christian. Гистиоцитоз Х (хронический)
- D76.1 Гемофагоцитарный лимфогистиоцитоз. Семейный гемофагоцитарный ретикулез. Гистиоцитозы из мононуклеарных фагоцитов, отличных от клеток Лангерганса, БДУ.
- D76.2 Гемофагоцитарный синдром, связанный с инфекцией
- D76.3 Другие гистиоцитозные синдромы. Ретикулогистиоцитома (гигантоклеточная). Синусный гистиоцитоз с массивной лимфаденопатией. Ксантогранулема
- D77 Другие нарушения крови и кроветворных органов при болезнях, классифицированных в других рубриках. Фиброз селезенки при шистосомозе [бильгарциозе] (B65.-)
Источник
Связанные заболевания и их лечение
Национальные рекомендации по лечению
Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Гистиоцитоз из клеток Лангерганса, Легочная эозинофильная гранулема.
Названия
Гистиоцитоз Х.
Синонимы диагноза
Гистиоцитоз из клеток Лангерганса, Легочная эозинофильная гранулема.
Описание
Гистиоцитоз X (икс), лангергансоклеточный гистиоцитоз — термин, обозначающий группу заболеваний с невыясненной до конца этиологией, при котором патологические иммунные клетки, называемые гистиоцитами и эозинофилы, активно размножаются, особенно в лёгких и костях, что вызывает формирование рубцовой ткани.
Симптомы
Различают 3 формы лангергансоклеточного гистиоцитоза:
• болезнь Хенда — Шюллера — Крисчена,.
• болезнь Абта — Леттерера — Сиве,.
• болезнь Таратынова или эозинофильная гранулёма.
Заболевания отличаются как по течению, так и по прогнозу, но так как это 3 формы одного заболевания, могут наблюдаться их взаимные переходы.
Болезнь Хенда — Шюллера — Крисчена может поражать детей в любом возрасте. Характерны дефекты костей черепа, таза, экзофтальм, несахарный диабет, ожирение, отставание в физическом развитии, гепатомегалия, лимфаденопатия, петехиальная сыпь, себорея, изменения в лёгких, стоматиты. К основному заболеванию может присоединиться вторичная инфекция.
Болезнь Абта — Леттерера — Сиве чаще встречается у детей раннего возраста. Развивается остро, с высокой лихорадкой, кожными высыпаниями в области грудины и позвоночника, гепатоспленомегалией (увеличение печени и селезенки), генерализованным увеличением лимфатических узлов, отитами, мастоидитами, поражением лёгких, поражением плоских костей.
Болезнь Таратынова наблюдается преимущественно у детей школьного возраста. Типичны: общая слабость, повышенная утомляемость, пониженный аппетит, боль в костях (поражаются как плоские, так и трубчатые кости), повышенная СОЭ, иногда эозинофилия. В ряде случаев болезнь протекает бессимптомно и заканчивается самопроизвольным излечением. На рентгенограммах костей обнаруживаются очаги деструкции, чаще округлой или овальной формы без зон склероза. В незначительном числе случаев клиническая картина болезни более яркая: несахарное мочеизнурение, экзофтальм, гепато- или гепатоспленомегалия, анемия, различные изменения кожи и.
Боль в грудной клетке. Глубокий сухой кашель. Жажда. Кашель. Лейкоцитоз. Лимфоцитопения. Молозивоподобные выделения из сосков. Моноцитоз. Ночная потливость у мужчин. Одышка. Понос (диарея). Постоянная жажда. Потливость. Рвота. Ретикулоцитоз. Судороги. Сухость во рту. Тошнота. Эозинофилия.
Причины
Этиология невыяснена. Предполагаются, что в основе гистиоцитоза X лежит некий иммунопатологический процесс, способствующий пролиферации гистиоцитов.
Лечение
Лечение, как правило, проводится в стационаре. При острых проявлениях назначают глюкокортикоиды в сочетании с цитостатиками, например, циклофосфамидом, хотя ни одно средство не является заведомо эффективным. Во всех случаях основная терапия сочетается с симптоматической.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: D58.0
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D55-D59 Гемолитические анемии / D58 Другие наследственные гемолитические анемии
Определение и общие сведения[править]
Микросфероцитарная гемолитическая анемия (синонимы: врожденный микросфероцитоз, болезнь Минковского-Шоффара, микроцитемия, сфероцитарная анемия) как самостоятельная болезнь стала известна после классических работ О. Минковского (1900) и Шоффара (Chauffard А.М., 1907).
Наследственный микросфероцитоз — это группа наследственных гемолитических анемий, характеризующихся появлением шаровидных эритроцитов (микросфероцитов) и обусловленных дефектом белков цитоскелета эритроцитов. В результате теряется часть мембраны эритроцита, уменьшается отношение площади поверхности к объему, и эритроцит превращается в микросфероцит.
Эпидемиология
В большинстве случаев заболевание наследуется аутосомно-доминантно (75% случаев). Распространенность его составляет 1 на 1000-4500. У 20% больных нарушения со стороны крови отсутствуют, что указывает либо на аутосомно-рецессивное наследование, либо на спонтанную мутацию (наблюдается сравнительно редко). Выраженность гемолиза и соответственно тяжесть заболевания весьма вариабельны. Чем более выражены проявления болезни, тем раньше ставится диагноз. Как правило, наследственный микросфероцитоз диагностируют в детстве, легкие формы с субклиническим течением — в зрелом возрасте, а иногда болезнь проявляется вскоре после рождения.
Этиология и патогенез[править]
Наследственный сфероцитоз вызван мутациями в одном из следующих генов: SPTA1 (1q21), SPTB (14q23.3), ANK1 (8p11.21), SLC4A1 (17q21.31) и EPB42 (15q15-q21), которые кодируют клеточные мембранные белки эритроцитов: спектрин 1 альфа-цепь, спектрин 1 бета-цепь, анкирин-1, транспортный белок полосы 3 и белок мембраны эритроцитов полосы 4.2, соответственно. Дефекты в этих белках приводят к потере сцепления мембраны эритроцитов и снижению площади поверхности мембраны, что приводит к образованию сферического слоя эритроцитов, снижению деформируемости и преждевременному разрушению в селезенке.
Спектрин — белок цитоскелета эритроцита; мутации его гена нарушают либо синтез цепей спектрина, либо самосборку его гетеродимеров. Спектрин представляет собой длинную фибриллярную молекулу длиной 200-260 нм и толщиной 2-3 нм. Его масса в клетке составляет около 30% массы мембранных белков. Молекула спектрина состоит из двух неидентичных субъединиц — альфа (240 000 Да) и бета (225 000 Да). Альфа- и бета-субъединицы ассоциируются в подвижные гетеродимеры. У каждого второго больного выявлены мутации гена анкирина — белка, соединяющего трансмембранный белок полосы 3 со спектрином. Недостаточность анкирина наследуется аутосомно-рецессивно или аутосомно-доминантно; аутосомно-рецессивное наследование встречается реже, но анемия при нем тяжелее. У четверти больных обнаружены мутации белка полосы 3. Это транспортный трансмембранный белок, его молекулярная масса около 100 000 Да. Белок носит название полосы 3, поскольку при электрофорезе в полиакриламидном геле он занимает соответствующее положение относительно других белков. Белок полосы 3 принимает участие в переносе кислорода из легких к тканям и углекислого газа из тканей к легким. Делеция гена белка полосы 3 делает мембрану эритроцита ригидной и защищает эритроциты от внедрения малярийных плазмодиев. Недостаточность этого белка наследуется аутосомно-доминантно и приводит к легкой анемии.
У большинства из оставшейся четверти больных выявлены мутации гена спектрина, нарушающие либо синтез цепей спектрина, либо самосборку его гетеродимеров. Недостаточность α -цепи спектрина наследуется аутосомно-доминантно и обычно протекает легко. Недостаточность β-цепи спектрина — тяжелое заболевание с аутосомно-рецессивным типом наследования. Видимо, неоднородность генетических нарушений обусловливает разнообразие в течении наследственного микросфероцитоза. Дефекты белков цитоскелета приводят к тому, что мембрана эритроцитов утрачивает стабильность и ее участки отщепляются. Эритроцит превращается в микросфероцит, неспособный к деформации. Микросфероциты не могут пройти через красную пульпу селезенки, в особенности протиснуться через щели в стенках ее синусов. Оказавшись в условиях гипоксии, в которых невозможно поддерживать метаболизм, микросфероциты теряют еще часть мембраны. В результате в крови появляется субпопуляция совершенно круглых эритроцитов.
Клинические проявления[править]
Основные проявления наследственного микросфероцитоза — анемия, желтуха, спленомегалия. Анемия обусловлена внутриклеточным распадом эритроцитов. Желтуха обусловлена непрямой гипербилирубинемией, бывает непостоянной и, как правило, слабее выражена в раннем детском возрасте. Из-за высокого содержания билирубина в желчи часто образуются пигментные желчные камни, в том числе и у детей. Спленомегалия наблюдается почти всегда. Во время системных инфекций интенсивность гемолиза может увеличиваться, что приводит к дальнейшему увеличению селезенки. При тяжелых формах наследственного микросфероцитоза у больных отмечаются деформации скелета: башенный череп, микрофтальмия, высокое верхнее нёбо, изменяется расположение зубов. У некоторых больных бывают укорочены мизинцы. Иногда возникают трофические язвы ног.
Выделяют четыре формы наследственного сфероцитоза: легкая (нормальный гемоглобин (Hb), ретикулоциты <3%, билирубин <17 мкмоль/л), мягкая (Hb 11-15 г/дл, ретикулоциты 3-6%, билирубин 17-34 мкмоль/л ), умеренная (Hb 8-12 г/дл, ретикулоциты> 6%, билирубин> 34 мкмоль/л) и тяжелая (Hb <8 г/дл, ретикулоциты> 10%, билирубин> 51 мкмоль/л).
Наследственный сфероцитоз: Диагностика[править]
1. Лабораторные данные и инструментальные исследования, необходимые для диагностики наследственной микросфероцитарной анемии: общий анализ крови — нормохромная анемия различной степени выраженности, появление микросфероцитов (эритроцитов уменьшенного диаметра шарообразной формы без просветления в центре) и ретикулоцитов в большом количестве. Анемия резко усиливается после активного гемолиза. Могут наблюдаться гемолитические кризы. Вне криза анемия умеренная, а при легком течении заболевания может отсутствовать. Микросфероциты характеризуются уменьшением диаметра (средний диаметр 4-6 мкм), увеличением их толщины и шарообразной формой. Чем тяжелее форма заболевания, тем большее количество микросфероцитов определяется в периферической крови. Количество лейкоцитов и тромбоцитов обычно нормальное. В период гемолитического криза наблюдаются лейкоцитоз и выраженный сдвиг лейкоцитарной формулы влево. СОЭ увеличивается только в периоде обострения заболевания, особенно во время гемолитического криза. Общий анализ мочи — определяется уробилинурия, а во время гемолитического криза — альбуминурия, микрогематурия. Биохимический анализ крови — повышено содержание билирубина преимущественно за счет неконъюгированного (непрямого) билирубина, во время гемолитического криза возможно повышение активности АЛТ ЛДГ, повышение содержания железа.
2. Осмотическая стойкость эритроцитов — отмечается снижение максимальной и минимальной осмотической стойкости эритроцитов. В норме минимальная стойкость составляет 0,44-0,48%, максимальная — 0,36-0,40% раствора натрия хлорида. При наследственной микросфероцитарной анемии гемолиз начинается при более высокой концентрации натрия хлорида: минимальная осмотическая резистентность понижена: 0,6-0,7%, а максимальная осмотическая резистентность повышена: 0,25-0,3%.
3. Миелограмма — в стернальном пунктате определяются характерные признаки гиперплазии красного кроветворного ростка — увеличение количества эритрокариоцитов. Гранулоцитарный и мегакариоцитарный ростки не изменены.
Отмечается значительное сокращение продолжительности жизни эритроцитов (по данным теста с радиоактивным хромом). В анализе кала возможно высокое содержание стеркобилина.
4. При УЗИ органов брюшной полости определяются увеличение селезенки, камни в желчном пузыре. При длительно существующем микросфероцитозе и частых обострениях возможно увеличение печени (вследствие нарушения оттока и застоя желчи).
Дифференциальный диагноз[править]
Дифференциальный диагноз включает наследственный эллиптоцитоз, наследственный стоматоцитоз, Южноазиатский овалоцитоз, дефицит глюкозо-6-фосфатдегидрогеназы, дефицит пируваткиназы, аутоиммунную гемолитическую анемию и альфа-талассемию.
Наследственный сфероцитоз: Лечение[править]
Лечение включает в себя купирование желтухи (фототерапия и обменное переливанием крови для предотвращения гипербилирубинемической энцефалопатии) и переливание эритромассы в случае тяжелой симптоматической анемии. Спленэктомия обычно приводит к исчезновению анемии и четкому улучшению гемолитических маркеров. Спленэктомия не показана пациентам с легкой формой, если нет жизненных показаний и ее желательно отсрочить до достижения возраста 6 лет. Лапароскопическая спленэктомия. Комбинированная спленэктомия и холецистэктомия могут быть полезны у пациентов с желчными камнями. Для предотвращения инфекций рекомендуется вакцинопрофилактика и назначение антибиотиков. Добавление фолатов рекомендуется, особенно после инфекционных осложнений. Уровни ферритина в сыворотке следует проверять ежегодно.
Прогноз
Прогноз варьируется и зависит от тяжести заболевания и сопутствующих осложнений.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Гематология [Электронный ресурс] : национальное руководство / под ред. О.А. Рукавицына — М. : ГЭОТАР-Медиа, 2015. — https://www.rosmedlib.ru/book/ISBN9785970433270.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Содержание
- Описание
- Дополнительные факты
- Классификация
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Профилактика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Агранулоцитоз.
Агранулоцитоз
Описание
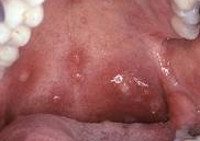
Агранулоцитоз. Клинико — гематологический синдром, в основе которого лежит резкое уменьшение или отсутствие нейтрофильных гранулоцитов среди клеточных элементов периферической крови. Агранулоцитоз сопровождается развитием инфекционных процессов, ангины, язвенного стоматита, пневмонии, геморрагических проявлений. Из осложнений часты сепсис, гепатит, медиастинит, перитонит. Первостепенное значение для диагностики агранулоцитоза имеет исследование гемограммы, пунктата костного мозга, обнаружение антинейтрофильных антител. Лечение направлено на устранение причин, вызвавших агранулоцитоз, предупреждение осложнений и восстановление кроветворения.
Дополнительные факты
Агранулоцитоз – изменение картины периферической крови, развивающееся при ряде самостоятельных заболеваний и характеризующееся снижением количества или исчезновением гранулоцитов. В гематологии под агранулоцитозом подразумевается уменьшение количества гранулоцитов в крови менее 0,75х109/л или общего числа лейкоцитов ниже 1х109/л. Врожденный агранулоцитоз встречается крайне редко; приобретенное состояние диагностируется с частотой 1 случай на 1200 человек. Женщины страдают агранулоцитозом в 2-3 раза чаще мужчин; обычно синдром выявляется в возрасте старше 40 лет. В настоящее время в связи с широким использованием в лечебной практике цитотоксической терапии, а также появлением большого количества новых фармакологических средств частота случаев агранулоцитоза значительно увеличилась.
Агранулоцитоз
Классификация
В первую очередь, агранулоцитозы подразделяются на врожденные и приобретенные. Последние могут являться самостоятельным патологическим состоянием или одним из проявлений другого синдрома. По ведущему патогенетическому фактору различают миелотоксический, иммунный гаптеновый и аутоиммунный агранулоцитоз. Также выделяют идиопатическую (генуинную) форму с неустановленной этиологией.
По особенностям клинического течения дифференцируют острые и рецидивирующие (хронические) агранулоцитозы. Тяжесть течения агранулоцитоза зависит от количества гранулоцитов в крови и может быть легкой (при уровне гранулоцитов 1,0–0,5х109/л), средней (при уровне менее 0,5х109/л) или тяжелой (при полном отсутствии гранулоцитов в крови).
На долю нейтрофильных гранулоцитов приходится до 50-75% всех белых кровяных телец. Среди них различают зрелые сегментоядерные (в норме 45-70%) и незрелые палочкоядерные нейтрофилы (в норме 1-6%). Состояние, характеризующееся повышением содержания нейтрофилов, носит название нейтрофилии. В случае понижения количества нейтрофилов говорят о нейтропении (гранулоцитопении), а в случае отсутствия – об агранулоцитозе. В организме нейтрофильные гранулоциты выполняют роль главного защитного фактора от инфекций (главным образом, микробных и грибковых). При внедрении инфекционного агента нейтрофилы мигрируют через стенку капилляров и устремляются в ткани к очагу инфекции, фагоцитируют и разрушают бактерии своими ферментами, активно формируя местный воспалительный ответ. При агранулоцитозе реакция организма на внедрение инфекционного возбудителя оказывается неэффективной, что может сопровождаться развитием фатальных септических осложнений.
Причины
Миелотоксический агранулоцитоз возникает вследствие подавления продукции клеток-предшественников миелопоэза в костном мозге. Одновременно в крови отмечается снижение уровня лимфоцитов, ретикулоцитов, тромбоцитов. Данный вид агранулоцитоза может развиваться при воздействии на организм ионизирующего излучения, цитостатических препаратов и других фармакологических средств (левомицетина, стрептомицина, гентамицина, пенициллина, колхицина, аминазина) и тд.
Иммунный агранулоцитоз связан с образованием в организме антител, действие которых обращено против собственных лейкоцитов. Возникновение гаптенового иммунного агранулоцитоза провоцирует прием сульфаниламидов, НПВС-производных пиразолона (амидопирина, анальгина, аспирина, бутадиона), препаратов для терапии туберкулеза, сахарного диабета, гельминтозов, которые выступают в роли гаптенов. Они способны образовывать комплексные соединения с белками крови или оболочками лейкоцитов, становясь антигенами, по отношению к которым организм начинает продуцировать антитела. Последние фиксируются на поверхности белых кровяных телец, вызывая их гибель.
В основе аутоиммунного агранулоцитоза лежит патологическая реакция иммунной системы, сопровождающаяся образованием антинейтрофильных антител. Такая разновидность агранулоцитоза встречается при аутоиммунном тиреоидите, ревматоидном артрите, системной красной волчанке и других коллагенозах. Агранулоцитоз, развивающийся при некоторых инфекционных заболеваниях (гриппе, инфекционном мононуклеозе, малярии, желтой лихорадке, брюшном тифе, вирусном гепатите, полиомиелите и тд ) также имеет иммунный характер. Выраженная нейтропения может сигнализировать о хроническом лимфолейкозе, апластической анемии, синдроме Фелти, а также протекать параллельно с тромбоцитопенией или гемолитической анемией. Врожденный агранулоцитоз является следствием генетических нарушений.
Патологические реакции, сопровождающие течение агранулоцитоза, в большинстве случаев представлены язвенно-некротическими изменениями кожи, слизистой оболочки полости рта и глотки, реже — конъюнктивальной полости, гортани, желудка. Некротические язвы могут возникать в слизистой кишечника, вызывая перфорацию кишечной стенки, развитие кишечных кровотечений. В стенке мочевого пузыря и влагалища. При микроскопии участков некроза обнаруживается отсутствие нейтрофильных гранулоцитов.
Симптомы
Клиника иммунного агранулоцитоза обычно развивается остро, в отличие от миелотоксического и аутоиммунного вариантов, при которых патологические симптомы возникают и прогрессируют постепенно. К ранними манифестным проявлениям агранулоцитоза относятся лихорадка (39-40°С), резкая слабость, бледность, потливость, артралгии. Характерны язвенно-некротический процессы слизистой оболочки рта и глотки (гингивиты, стоматиты, фарингиты, ангины), некротизация язычка, мягкого и твердого нёба. Данные изменения сопровождаются саливацией, болью в горле, дисфагией, спазмом жевательной мускулатуры. Отмечается регионарный лимфаденит, умеренное увеличение печени и селезенки.
Для миелотоксического агранулоцитоза типично возникновение умеренно выраженного геморрагического синдрома, проявляющегося кровоточивостью десен, носовыми кровотечениями, образованием синяков и гематом, гематурией. При поражении кишечника развивается некротическая энтеропатия, проявлениями которой служат схваткообразные боли в животе, диарея, вздутие живота. При тяжелой форме возможны осложнения в виде прободения кишечника, перитонита.
Нейтрофилез. Понос (диарея). Потливость.
Диагностика
Группу потенциального риска по развитию агранулоцитоза составляют пациенты, перенесшие тяжелое инфекционное заболевание, получающие лучевую, цитотоксическую или иную лекарственную терапию, страдающие коллагенозами. Из клинических данных диагностическое значение представляет сочетание гипертермии, язвенно-некротических поражений видимых слизистых и геморрагических проявлений.
Наиболее важным для подтверждения агранулоцитоза является исследование общего анализа крови и пункция костного мозга. Картина периферической крови характеризуется лейкопенией (1-2х109/л), гранулоцитопенией (менее 0,75х109/л) или агранулоцитозом, умеренной анемией, при тяжелых степенях – тромбоцитопенией. При исследовании миелограммы выявляется уменьшение количества миелокариоцитов, снижение числа и нарушение созревания клеток нейтрофильного ростка, наличие большого количества плазматических клеток и мегакариоцитов. Для подтверждения аутоиммунного характера агранулоцитоза производится определение антинейтрофильных антител.
Всем пациентам с агранулоцитозом показано проведение рентгенографии легких, повторные исследования крови на стерильность, исследование биохимического анализа крови, консультация стоматолога и отоларинголога.
Дифференциальная диагностика
Дифференцировать агранулоцитоз необходимо от острого лейкоза, гипопластической анемии. Также необходимо исключение ВИЧ-статуса.
Профилактика
Пациенты с верифицированным агранулоцитозом должны быть госпитализированы в отделении гематологии. Больные помещаются в палату-изолятор с асептическими условиями, где проводится регулярное кварцевание, ограничивается посещение, медицинский персонал работает только в шапочках, масках и бахилах. Эти меры направлены на предупреждение инфекционных осложнений. В случае развития некротической энтеропатии осуществляется перевод больного на парентеральное питание. Пациентам с агранулоцитозом необходим тщательный уход за полостью рта (частые полоскания рта антисептическими растворами, смазывание слизистых оболочек).
Лечение
Терапия агранулоцитоза начинается с устранения этиологического фактора (отмены миелотоксических препаратов и химических веществ ). Для профилактики гнойной инфекции назначаются неабсорбируемые антибиотики, противогрибковые препараты. Показано внутривенное введение иммуноглобулина и антистафилококковой плазмы, трансфузии лейкоцитарной массы, при геморрагическом синдроме — тромбоцитарной массы. При иммунном и аутоиммунном характере агранулоцитоза назначаются глюкортикоиды в высоких дозах. При наличии в крови ЦИК и антител проводится плазмаферез. В комплексном лечении агранулоцитоза используются стимуляторы лейкопоэза.
Прогноз
Профилактика агранулоцитоза, главным образом, заключается в проведении тщательного гематологического контроля во время курса лечения миелотоксическими препаратами, исключении повторного приема лекарств, ранее вызвавших у больного явления иммунного агранулоцитоза. Неблагоприятный прогноз наблюдается при развитии тяжелых септических осложнений, повторном развитии гаптеновых агранулоцитозов.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник