Растяжимость кожи при синдроме элерса данлоса

В этой статье мы расскажем о такой патологии, как синдром Элерса-Данлоса, что это такое, рассмотрим симптомы (фото больных), причины возникновения и метод терапии.
Что такое синдром Элерса-Данлоса
Синдром Элерса-Данлоса – это наследственное заболевание, спровоцированное дефектом синтеза коллагена. При такой заболевании наблюдается неправильное развитие соединительных тканей и гиперэластичность кожи.
Синдром имеет несколько типов, которые могут характеризоваться неестественной растяжимостью кожи, чрезмерной способностью суставов разгибаться в разные стороны, косоглазием, деформацией скелета.
Болезнь затрагивает большое количество систем организма и представляет интерес не только с точки зрения генетики, но и кардиологии, ортопедии, стоматологии, офтальмологии и других медицинских дисциплин.
Так же синдром Элерса-Данлоса отличается сложностью в диагностике из-за наличия легких форм недуга, поэтому нельзя с точностью оценить его распространенность в мире.
Эпидемиология
Точная распространенность и годовая заболеваемость неизвестны, оценки распространенности варьируются от 1/5 до 000-1/20000. Из-за клинической изменчивости эти оценки могут быть слишком низкими. Больше всего болезнь затрагивает – женский пол.
Причины синдрома Элерса-Данлоса
Синдром Элерса-Данлоса имеет несколько вариантов патологии, отличающихся друг от друга по типу наследования. Но их объединяет то , что в их основе лежит структурное либо количественное нарушение белка коллагена. В нынешнее время не для каждого типа заболевания найдены молекулярные механизмы синдрома.
Для первого типа синдрома характерна пониженная активность тех клеток соединительной ткани, отвечающие за синтез межклеточного матрикса. Так же происходит увеличение синтеза протеогликанов, а еще отсутствие ферментов, отвечающих за стабильное воссоздание белков коллагена.
При четвертом типе синдром Элерса-Данлоса наблюдается дефицит коллагена 3 типа. Шестой тип характеризуется недостатком биологического катализатора лизилгидроксилазы, который принимает участие в гидроксилировании лизина в молекулах проколлагена. Седьмой тип заболевания возникает вследствие изменения проколлагена первого типа, который становиться коллагеном.
Если охарактеризовать заболевание в целом, в различных формах, то заболевание возникает вследствие снижения плотности и расстройство ориентации коллагеновых волокон, утончение соединительного отдела кожи, расширение кровеносных сосудов и большую видимость их на поверхности тела.
Симптомы синдрома Элерса-Данлоса
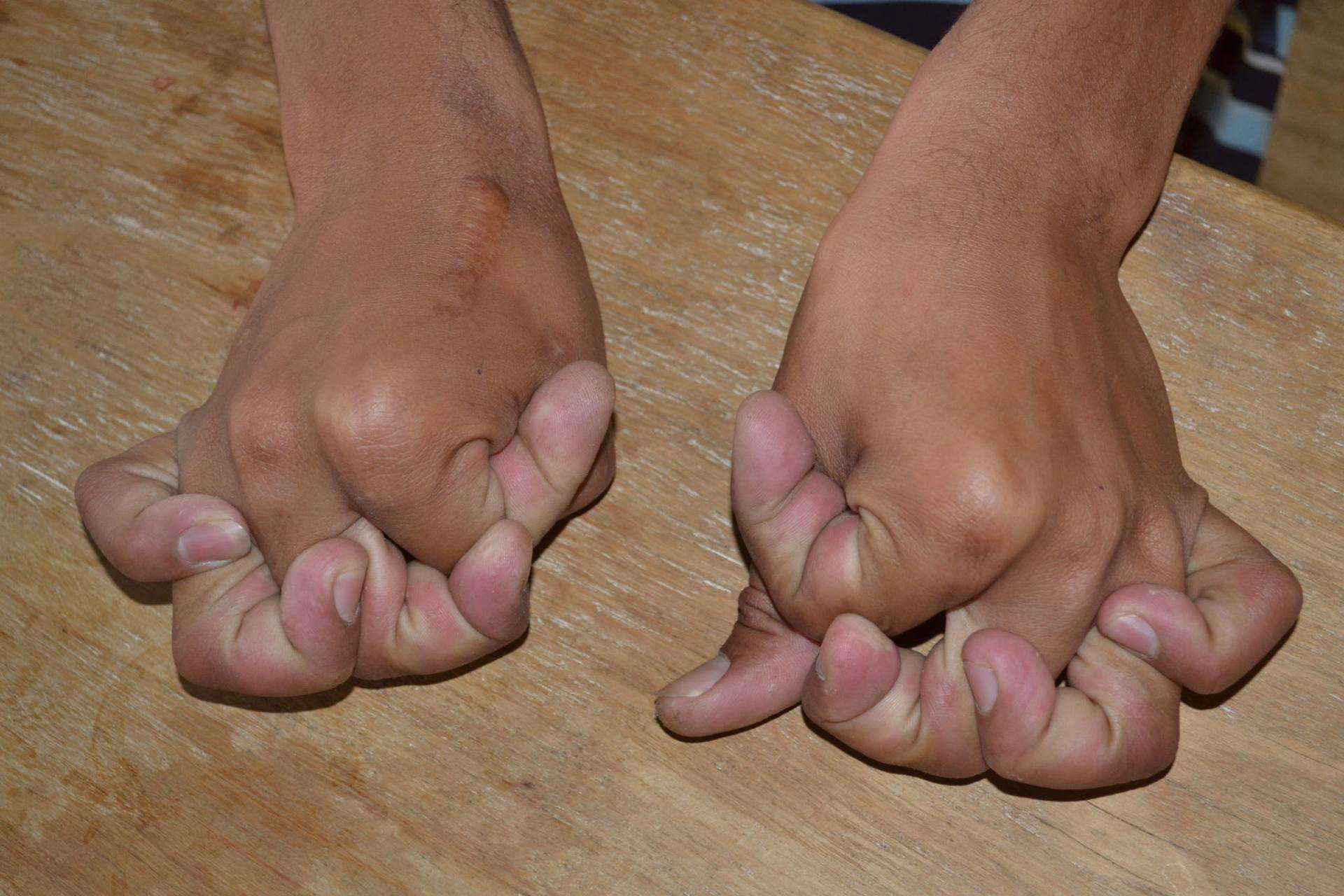
Симптомы могут возникать в любом возрасте, но их трудно диагностировать у маленьких детей из-за повышенной гипервозбудимости суставов. Клинические проявления очень разные. Первичное проявление – это это гиперэластичность различных суставов и аномальная эластичность кожного покрова (см. фото выше).
На ощупь кожа больного нежная, но на ступнях и ладонях присутствуют многочисленные морщины. При синдроме Элерса-Данлоса неестественная эластичность кожи наблюдается с ранних лет жизни человека, так же с возрастом характерно снижение такого отклонения.
Повреждения на коже людей, подверженных синдрому, ощущаются сильней и заживают медленно, а на их месте образуются следы в виде шрамов и опухолей.
Кроме гиперэластичности кожи у подверженных синдрому Элерса-Данлоса наблюдается сильная гиперэластичность суставов, как отдельной части тела, так и всего организма.
Суставы становятся невероятно гибкими и гнуться под неестественными им углами с того момента, как человек начинает учиться ходить, что нередко приводит к травмам. С возрастом ненормальная способность суставов к сгибанию ослабевает.
Повреждение скелета при синдроме представляет собой деформацию грудной клетки в виде дуги или полукруга.
В результате заболевания возникает сколиоз, косолапость. Со скелетом нарушается и расположение внутренних органов, опущение, наличие различных видов грыж, дивертикулез кишечника, пневмоторакс, ввиду нарушения целостности плевры.
Наличие синдрома ведет к патологиям зрения, среди которых встречаются:
- миопия;
- спонтанная отслойка сетчатки;
- косоглазие;
- разрыв глазного яблока и роговицы.
Что касается кардиологии, то у детей с синдромом Элерса-Данлоса возникают частые кровотечения и гематомы.
Не исключен порок сердца, варикоз, пролапс митрального клапана и аневризмы сосудов головного мозга. Нарушений умственного развития детей с данным заболеванием обычно не наблюдается.
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Диагностика синдрома Элерса-Данлоса
Диагностику синдрома Элерса-Данлоса лучше доверять опытному человеку, специализирующемуся именно в области этого заболевания. Малознакомому с Элерсом-Данлосом врачу будет сложно выявить недуг и тем более его особенности. Это требование связанно с разнообразием индивидуальных проявлений такого заболевания у каждого пациента. Существует множество разновидностей болезни, но есть характерные черты ее проявления.
Неестественная эластичность кожного покрова выявляется путем оттягивания кожи до того момента, пока не почувствуется сопротивление. Участки для осуществления проверки следует выбирать максимально нейтральные, где нормальная оттягиваемость кожи превышает полтора сантиметра.
Среди характерных для болезни синдромов присутствует гипермобильность суставов. Такой симптом определяется по шкале Бейтона, где 5 баллов означает отклонение от нормы. Этот симптом, с возрастом, наблюдается в меньшей степени.
О хрупкости суставных соединений свидетельствуют спонтанно возникающие синяки. Вместе с этим возможны долговременные кровотечения.
Даже при совсем слабых внешних воздействиях появляются различные травмы , которые оставляют атрофические следы ранений в виде шрамов и расширяются с течением времени. С ами ранения заживают дольше, чем у здоровых людей.
Характерной патологией синдрома Элерса-Данлоса является пролапс митрального клапана. Этот недуг выявляется проведением УЗИ сердца.
Постоянная боль в области суставов и конечностей тела – еще одно проявление синдрома Элерса-Данлоса. При таком признаке рентген может не выявить отклонений, а пациент не всегда понимает, из какой части тела исходят болевые ощущения.
Из шкалы Брайтона берутся формальные критерии для того, чтобы диагностировать синдром Элерса-Данлоса. Первоначально функцией шкалы Бейтона была диагностика гипермобильности в суставах, но современные опыты показывают, что синдром гипермобильности суставов и повышенная сгибаемость суставов при Элерсе-Данлосе, – это аналогичные заболевания.
Шкала Бейтона
Основные критерии:
- 4+ баллов по шкале Бейтона выявленных в настоящее время, либо раньше;
- артралгия, длящаяся на протяжении 3 месяцев и более в 4 или более суставах.
Второстепенные критерии:
- 1-3 Балла по шкале Бейтона ( и 0 если вы старше 50-ти);
- артралгия в 1, 2 или 3 суставах с присутствием болезненных ощущений в спине;
- вывихи в одном и более суставах, либо в одном суставе несколько раз;
- более 3 воспалений мягких тканей, ревматизм;
- марфаноидный тип внешности (чрезмерная худощавость, высокий рост, арахнодактилия);
- необычные шрамы и ненормальная растяжимость и тонкость кожи;
- растянутая кожа век или миопия;
- ректальный пролапс, варикозное расширение вен, пролапс матки, грыжа.
Для того чтобы поставить диагноз “синдром Элерса-Данлоса” нужно выявить оба основных критерия, либо 1 из основных и 2 любых второстепенных, для постановки диагноза подойдет и наличие 4 второстепенных критериев.
Когда у вашего первостепенного родственника выявлен данный синдром, вам достаточно будет определить наличие 2 второстепенных критериев.
Лечение синдрома Элерса-Данлоса
Для эффективного устранения синдрома Элерса-Данлоса специфическая терапия до сих пор не разработана, но существуют методы лечения симптомов данной болезни.
Медикаментозная терапия
Для того, чтобы стабилизировать работу сердечно-сосудистой и нервной системы, а так же привести в норму работу суставов, опорно-двигательный аппарат, целостность и эластичность кожного покрова используют несколько вариантов лечения:
- лекарственные препараты в виде аскорбиновой кислоты, А, Е, В витаминов;
- комплексы минералов;
- инъекции соматотропного гормона для стимуляции роста;
- стимулирующие обмен веществ и регенерацию кожного покрова метаболические средства, такие как карнитин-хлорид;
- для стимуляции мозговой деятельности применяются нейрометаболические стимуляторы;
- для поддержания целостности скелета и соединительных тканей используются остеокеа или остеогенон;
- заживлению кожи способствуют такие препараты, как инозин, АТФ, коэнзим Q10.
- используется принимающий участие в синтезе хрящевых и в соединительных тканей глюкозамин.
Хирургическое вмешательство
К хирургическому вмешательству при синдроме Элерса-Данлоса стоит прибегать, только если возникают осложнения, угрожающие жизни. Перед хирургическими операциями должны быть проведены тщательные инвазивные диагностические процедуры для того чтобы оценить степень угрозы и необходимость хирургического вмешательства.
Среди хирургического лечения данного синдрома может быть необходимым: удаление псевдоопухолей, коррекция ВПС, реконструкция грудной стенки и т.д.
Дополнительные и альтернативные методы лечения, в том числе и в домашних условия
Среди процедур назначаются:
- лечебная физкультура и массаж
- рефлексотерапия (воздействие на биологически активные точки организма, отвечающие за работу различных его систем)
- различные физиотерапевтические процедуры (к примеру, лазерная акупунктура)
Также назначается диета с повышенным потреблением белка. Такая диета содержит потребление костных бульонов и заливных блюд.
Прогноз для больных
Повышенного риска ранней смертности нет, однако, в связи с нестабильностью суставов, хроническим и острым болевым синдромом, и симптомами проявляющееся вне костно-мышечной системы, качество жизни больного серьезно ухудшается.
Как уже говорилось выше, специального лечения нет. Необходимо проходить индивидуализированные вспомогательные и симптоматические методы лечения включающие физиотерапию, реабилитацию, прием анальгетиков и соответствующую терапию внесуставных симптомов. Хирургические меры следует рассматривать с умом.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 26 июля 2015;
проверки требуют 20 правок.
Синдром Э́лерса — Данло́са (синдром Э́лерса — Данло́[2], син. Э-Д; англ. Ehlers-Danlos Syndrome, «гиперэластичность кожи» («Cutis hyperelastica»[3]), несовершенный десмогенез, несовершенный десмогенез Русакова, синдром Черногубова — Элерса — Данлоса) — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром назван в честь двух дерматологов, идентифицировавших его в начале XX века: Эдварда Элерса[en] (1863—1937) из Дании и Анри-Александра Данлоса[fr] (1844—1912) из Франции[4].
Симптоматика[править | править код]
Человек с син. Э-Д демонстрирует гиперподвижность суставов. |
Симптомы сильно варьируются в зависимости от типа болезни. Болезнь, как правило, поражает суставы, кожу и кровеносные сосуды, с симптомами, такими как свободные (плохо прикреплённые), сильно гнущиеся суставы; гладкая или эластичная, легко повреждающаяся кожа; неправильное заживление ран и формирование шрамов; маленькие и хрупкие кровеносные сосуды. Все формы затрагивают суставы, вызывая гиперподвижность, они выходят за пределы нормального диапазона движений.[5] В результате люди с «син. Э-Д» более подвержены различным травмам таким как: вывихи, подвывихи, растяжения связок, деформация и иногда разрыв мягких тканей. Так как синдром часто не диагностируется, некоторые случаи принимаются за жестокое обращение с детьми.[6]
Классификация[править | править код]
В прошлом было более десяти общепризнанных типов «син. Э-Д». В 1997 исследователи предложили более простую классификацию, которая сократила число главных типов до шести и дала им описательные имена.[7] Другие типы условно могут существовать, но о них сообщается только в отдельных семьях или они плохо характеризованы. За исключением типа гиперподвижности, были идентифицированы и отождествлены отдельные мутации — они могут быть точно определены генетическим анализом. Это особенно важно из-за значительной вариативности признаков в индивидуальном рассмотрении, из-за чего может быть перепутана классификация только на симптоматической основе.
По распространенности среди населения:
| Имя | Номер | Описание | OMIM | Ген(ы) |
| Гиперподвижность (Hypermobility) | тип 3 | Поражает 1 человека на 10 000 — 15 000, вызван аутосомным доминирующим механизмом. Возникает в результате мутации любого из двух генов, которые вызывают Сосудистый тип и син. Э-Д с дефицитом тенасцина-X, соответственно. Проявляется в гиперподвижности суставов; поражение кожи выражено меньше. Характерны хронические мышечно-скелетные боли. | 130020 | COL3A1, TNXB |
| Классический (Classical) | тип 1 и 2 | Поражает приблизительно от 2 до 5 человек на 100 000. Затрагивает коллаген типа V, также коллаген типа I. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Сосудистый (Vascular) | тип 4 | Поражает приблизительно 1 человека на 100 000. Вызван аутосомно-доминантным дефектом в синтезе коллагена типа III | 130050 | COL3A1 |
| Кифосколиоз (Kyphoscoliosis) | тип 6 | Аутосомно-доминантный дефект, вызывающий недостаток фермента, называемого Лизин Гидролаза[1]. Очень редок; описано немногим более 60 случаев. | 225400, 229200 | PLOD1 |
| Артрохалазия (Arthrochalasis) | типы 7A и B | Поражает коллаген типа I. Крайне редок, описано всего около 30 случаев. | 130060 | COL1A1, COL1A2 |
| Дерматоспараксис (Dermatosparaxis) | тип 7C | Также крайне редок, описано около 10 случаев. | 225410 | ADAMTS2 |
Тип гиперподвижность[править | править код]
Прежний тип 3, встречается у 1 человека на 10 000 — 15 000, что делает его самым распространённым вариантом болезни. Признаки и симптомы могут быть не диагностированы (не признаны) врачами или, как правило, ошибочно диагностированы как фибромиалгия и обычно больным не ставят диагноз, пока не проявятся серьёзные осложнения. Диагностика осуществляется, в основном, на клинических наблюдениях.
Основные признаки и симптомы включают в себя:
- Свободные, нестабильные суставы, подверженные: растяжениям, вывихам, подвывихам (частичный вывих), переразгибанию суставов
- Плоскостопие
- Высокое и узкое нёбо
- Лёгкие кровоподтёки
- Легко повреждающаяся бархатно-гладкая кожа
- Раннее начало остеопороза (обычно проявляется в середине 30 лет)
- Поражение сердца: в некоторой степени en:Dysautonomia или приобретённый порок сердца (такой как пролапс митрального клапана, что создаёт повышенный риск инфекции (эндокардит) во время операций (также возможно развитие до крайне опасной для жизни степени при пролапсе митрального клапана).[8]
Другие симптомы и осложнения могут включать в себя:
- Низкая плотность костей (остеопения) — предшественник остеопороза
- Мышечная слабость, часто усугубляется холодной погодой
- Деформации позвоночника, такие как: сколиоз (искривление позвоночника), кифоз (горб в грудном отделе), en:Tethered spinal cord syndrome, базилярная инвагинация (cranial settling), а также Мальформация Арнольда — Киари (поражение продолговатого мозга, мозжечка выраженные затылочными болями, нарушениями глотания, атаксией и другими симптомами)[9]
- Функциональные расстройства кишечника (функциональный гастрит, синдром раздражённого кишечника)
- Сдавление нервов (синдром запястного канала, парестезия, невралгия тройничного нерва)
- Болезнь Рейно
- Миалгия (боль в мышцах) и артралгия
- Чрезмерная усталость
- Преждевременный разрыв амниона (выкидыш) во время беременности en:Premature rupture of membranes
- Младенцы с гипервподвижностью суставов имеют слабый мышечный тонус (мышечная гипотония), который может задержать развитие таких моторных навыков как самостоятельное присаживание, вставание и хождение.
Боль, сопутствующая этому состоянию, является серьёзным осложнением.
Классический тип[править | править код]
При старой системе классификации он был разделён на два типа: тип I (тяжёлый) и тип II (умеренный). Поражает приблизительно от 2 до 5 человек на 100 000, и является вторым по распространённости. Поражает коллаген V и I типа. Важные симптомы затрагивают кожу и суставы. Больные как правило проявляют:
- гладкая, сильно эластичная, легко ранимая кожа
- уродливые или необычайно обширные шрамы, особенно на лбу, коленях, локтях и подбородке
- гиперподвижные суставы имеют тенденцию к вывихам, растяжению связок и подвывихам (обычно в коленной чашечке, в плече, в пястно-фаланговом суставе en:Metacarpophalangeal joint, и в височно-челюстном суставе
- Из-за сниженного мышечного тонуса, у младенцев может быть нарушено развитие моторных навыков
- Дети могут иметь склонность к развитию грыжи или смешению любого внутреннего органа.
В настоящее время не существует определённого теста для диагностики этого типа синдрома. И ДНК анализ и биохимические исследования используются для выявления пораженных болезнью. В некоторых случаях биопсия кожи была признана полезной при постановке диагноза. К сожалению эти тесты не достаточно надёжны, чтобы выявить всех больных. Если в семье есть несколько больных членов, то можно провести внутриутробную ДНК диагностику.
Сосудистый тип[править | править код]
Поражает приблизительно 1 человека на 100 000, вызван аутосомным доминантным дефектом в синтезе коллагена типа III. В старой системе классификации имел номер IV, этот тип является самой опасной разновидностью синдрома. Проведенные исследования определяют ожидаемую продолжительность жизни примерно в 48 лет. Тем не менее, эта цифра вероятно искажена и основана на факте, что этот тип (как все остальные типы синдрома) значительно не выявлен, и высокая пропорция смертельных случаев вызвана посмертным диагностированием. Повышение осведомлённости среди врачей и населения может помочь сделать эту цифру более точной, и сократить число преждевременных смертей.
Признаки и симптомы:
- Гиперподвижность, наиболее очевидна на пальцах рук и ног
- Хрупкие стенки сосудов оболочек органов и нежной кожи, имеют склонность к разрыву или образованию аневризмы
- Больные как правило имеют тонкую, бледную и прозрачную кожу (можно видеть вены на груди)
- Артериальная/кишечная/маточная хрупкость или трещины, разрывы
- обширные кровоподтёки
- Некоторые пациенты выражают характерные черты лица (большие глаза, маленький подбородок, тонкий нос и губы, мягкие уши) и имеют маленький рост
В результате возможности маточного разрыва, беременность может оказаться опасной для жизни. Доступно лабораторное тестирование. Кожная биопсия может служить доказательством аномальной структуры коллагена. Этот биомеханический анализ выявляет более 95 % случаев. Лабораторное тестирование рекомендуется лицам имеющим два или более значительных симптома. ДНК анализ может выявить изменения в пределах гена COL3A1. Эта информация может помочь при предродовой генетической консультации, когда один из родителей был диагностирован и известна его генетическая мутация или был продемонстрирован биомеханический дефект.
Тип кифосколиоз[править | править код]
В прежней классификации тип VI. Он очень редок, обнаружено немногим более 60 случаев, передаётся аутосомно-рецессивным механизмом. Основным симптом является общая нестабильность (непрочность) суставов. У младенцев наблюдается слабый мышечный тонус, задержка в развитии моторных навыков, прогрессирующее в течение жизни ненормальное искривление позвоночника сколиоз, при котором обычно больные не могут ходить к 20 годам. Легко ранимые глаза и кожа, также вероятна уязвимость кровеносных сосудов. Наблюдаются: спонтанная отслойка сетчатки, кровоизлияния в стекловидное тело, разрывы глазного яблока и роговицы, склер. Также у костей может быть снижена плотность.
Существует четыре основных медицинских критерия при диагностике этого типа. Это гиперподвижные суставы, слабый мышечный тонус у новорождённых, прогрессирующий с рождения сколиоз и хрупкие (уязвимые) глаза, что может придать голубоватый оттенок склерам или вызвать разрыв глаза.
Вызван изменениями в хромосоме 1 гена PLOD, который кодирует фермент лизил-гидролазу. Возможен лабораторный тест, в нём измеряется содержание в моче hydroxylysyl pryridinoline’а. Он крайне чувствителен и специфичен для данного типа синдрома, рекомендуется младенцам с тремя и более основными симптомами. Внутриутробный анализ применяется, если известно о существующем риске: диагностированы больные члены семьи, имеющие положительные результаты тестирования. Вышеупомянутый амниоцентез может быть выполнен, если зародышевые клетки берутся из амниотической жидкости и измерена активность фермента.
Артрохалазия[править | править код]
Характеризуется дефектом коллагена I типа за счет генов COL1.
При этом варианте ребенок может рождаться с врожденным вывихом бедра. Наследуется аутосомно – доминантно. Описано только около 30 случаев.[10]
Симптомы:
- Тяжелая генерализованная гипермобильность суставов с повторными вывихами (подвывихами)
- Гиперэластичная кожа;
- Атрофические шрамы;
- Мышечная гипотония;
- Кифосколиоз;
- Остеопения.[10]
Дерматоспараксис[править | править код]
Имеется дефектный ген ADAMTS2, о котором было сообщено в 10 случаях по всему миру.
Наследуется аутосомно – рецессивно.
Симптомы:
- Чрезвычайно хрупкая кожа, склонная к появлению синяков
- Раны и ссадины тяжело и долго заживают, образуя впоследствии атрофические шрамы
- Кожа на ощупь мягкая и податливая, ее слишком много и она образует складки
- Преждевременный разрыв плодных оболочек, грыжи (пупочные, паховые)[10]
Диагностика[править | править код]
Основана на данных:
- Анамнеза заболевания
- Обследования
- Биопсии кожи
- Молекулярно-генетических исследований
Дополнительно проводят:
- УЗИ внутренних органов
- Офтальмологические обследования
- Эхокардиограмму[10]
Лечение[править | править код]
Симптоматическое:
Для стабилизации деятельности сердечно-сосудистой и нервной системы, нормализации процессов в опорно-двигательном аппарате и коже используются:
- гемостатическая терапия — аскорбиновая кислота, этамзилат, антифибринолигики;
- витаминные препараты (витамины А, Е, В, С);
- препараты, стимулирующие обмен веществ и регенерацию;
- метаболические средства (карнитин-хлорид);
- минеральные комплексы;
- препараты, стимулирующие рост (инъекции соматотропного гормона);
- нейрометаболические стимуляторы, стимулирующие умственную деятельность.[10]
Профилактика[править | править код]
Основными направлениями профилактики неблагоприятных проявлений СЭД являются:
- правильно подобранная физическая нагрузка;
- предотвращение дислокаций;
- профилактические курсы лечения у офтальмолога, стоматолога;
- удаление псевдоопухолей;
- лечение патологии сердца, глаз.
См. также[править | править код]
Дисплазия соединительной ткани
Ссылки[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ В русскоязычной литературе распространены оба названия, «Синдром Элерса — Данло́са» и «Синдром Элерса — Данло́». Большая медицинская энциклопедия (том 29, 1998), Геномная энциклопедия человека (1991), Малая медицинская энциклопедия (Т. 1, 1991) дают написание «Элерса-Данлоса».
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. Page 512. ISBN 0-7216-2921-0.
- ↑ «Uncovered: How U.S. Health System Can Fail Even the Insured — A Woman Endures 16-Month Odyssey To Get a Diagnosis», John Carreyrou, Wall Street Journal, November 16, 2007
- ↑ Lawrence E.J. The clinical presentation of Ehlers-Danlos syndrome (англ.) // Advances in Neonatal Care (англ.)русск. : journal. — 2005. — Vol. 5, no. 6. — P. 301—314. — doi:10.1016/j.adnc.2005.09.006. — PMID 16338669.
- ↑ The Press Enterprise, Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries Архивировано 28 февраля 2009 года., 2008-04-03
- ↑ Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) (англ.) // American Journal of Medical Genetics (англ.)русск. : journal. — 1998. — Vol. 77, no. 1. — P. 31—7. — doi:10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O. — PMID 9557891.
- ↑ Ehlers-Danlos Syndrome, Hypermobility Type — GeneReviews — NCBI Bookshelf
- ↑ Sorry, but we cannot find this page. Дата обращения 10 апреля 2013.
- ↑ 1 2 3 4 5 Румянцев А.Г. Федеральные клинические рекомендации по диагностике и лечению синдрома Элерса-Данло у детей (2015).
Источник