Птицеголовая карликовость или синдром секкеля

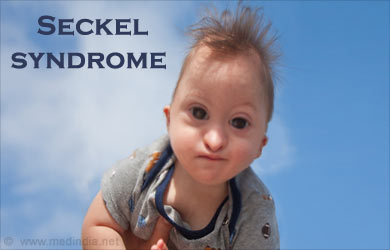
Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Секкеля — лечение в Италии
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) — редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птицеголовость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефалия» принадлежат Н.Зеске в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Синдром Секкеля — редкий синдром, характеризующийся тяжелой низкорослостью, низким весом при рождении, очень маленькой головой (микроцефалия), большим лбом, большими глазами, низкими ушами, большим носом и небольшим подбородком. Дефекты костей рук и ног, вывихи локтевых суставов и бедер, неспособность выпрямлять колени и крипторхизм, также часто встречаются у детей с этим синдромом.
Хромосомная нестабильность и недопроизводство всех типов клеток крови (панцитопения) отмечаются у некоторых пациентов. Это заболевание является генетическим. Оно наследуется по аутосомно-рецессивному типу. Синдром Секкеля является неоднородным генетическим заболеванием, которое может быть связано с мутациями в генах, расположенных на хромосомах 3 и 18.
Следует отличать синдрос Секкеля от другиз заболеваний с низкорослостью. Для этого проводится дифференциальная диагностика при характерных дополнительных симптомах.
Низкая масса тела при рождении в случае доношенной беременности, внутриутробная задержка роста. Этот частый симптом свидетельствует о наличии генетически обусловленного снижения потенции роста тканей, недостаточности плаценты или же нарушений роста во внутриутробном периоде токсического или воспалительного генеза. Если отсутствует дополнительная симптоматика и вместе с тем имеющуюся клиническую картину нельзя отнести к какому-либо определенному заболеванию, говорят о примордиальной низкорослости. При отсутствии других симптомов последняя описывается как семейное заболевание.
В спорадических случаях следует думать о необратимом внутриутробном повреждении редупликационного потенциала клеток неизвестным патогенным фактором. При чисто примордиальной низкорослости дифференцировка скелета соответствует хронологическому возрасту.
- а) Примордиальная низкорослость: синдром Рассела — Сильвера.
- б) Синдром Секкеля: гетерогенная группа примордиальных карликов с микроцефалией, большим носом, скошенным подбородком и интеллектуальным недоразвитием.
- в) Прогерия (синдром Гатчинсона-Джилфорда): замедление роста и развитие начиная со 2-3-го года жизни, преждевременное старение, атрофия кожи, выпадение волос.
- г) Синдром Блума: телеангиэктатическая эритема щек, долихоцефалия.
- д) Синдром Корнелии де Ланге; умственное недоразвитие, густые, сросшиеся над переносицей брови, микроцефалия, усиленное оволосение тела.
- е) Лепрехаунизм (синдром Донохуа): отсутствие или недоразвитие подкожной жировой клетчатки, гипертрофия клитора или полового члена, усиленное оволосение.
- ж) Синдром Фанкони: панцитопения, гипоплазия I пальца руки или нарушение его закладки, пигментные аномалии.
- з) Семейная, или полигенная, низкорослость, при которой в большинстве случаев наблюдается только легкая задержка роста во внутриутробном периоде.
- и) Трисомия 18.
Очень короткие конечности (увеличенное отношение верхнего сегмента к нижнему). К этой группе прежде всего относятся заболевания скелета, обусловленные поражением эпифизов и метафизов. При дифференциальной диагностике обязательно следует подумать об атиреозе или тяжелом гипотиреозе (особенно учитывая возможности их лечения). Фосфатный диабет также может сопровождаться легкой диспропорциональной низкорослостью с преобладанием верхнего сегмента. Пропорции могут существенно меняться в процессе роста. Здесь представлены сведения о пациентах старшего детского и юношеского возраста в соответствии с обобщением Спрангера.
- а) Ахондроплазия (хондродистрофия): относительно большая голова с провалившимся основанием носа, особенное укорочение предплечий и голеней, кифоз, типичные рентгенологические находки.
- б) Гипохондроплазия: слабо выраженная диспропорциональная низкорослость, переразгибание суставов, люмбальный лордоз, типичные рентгенологические данные.
- в) Псевдоахондроплазия: пропорции, как и при ахондроплазии, но с нормальным формированием мозгового и лицевого черепа, относительно позднее по сравнению с ахондроплазией проявление заболевания, отсутствие типичных рентгенологических симптомов со стороны таза.
- г) Синдром Эллиса-Ван-Кревельда: порок сердца, дистрофия ногтей и пальцев стоп, эписпадия.
- д) Синдром Маркезани: изменения хрусталиков, как при синдроме Марфана (дрожание хрусталиков, может быть шаровидный хрусталик), брахидактилия, возможны легкие контрактуры.
- е) Метафизарная хондродисплазия, тип Шмидта: укорочение трубчатых костей (больше проксимальных), грибовидно вздутые эпифизы с размытыми очертаниями.
- ж) Метафизарная хондродисплазия, тип Мак-Кусика: редкие тонкие волосы, брови и ресницы, переразгибание суставов, типичная рентгенологическая картина.
- з) Метафизарная хондродисплазия, тип Енсена: утолщенные суставы с костным ограничением подвижности, формирование косолапости стоп, типичная рентгенологическая картина.
- и) Дисхондростеоз (синдром Лери — Вейля): укорочение предплечья и лучевой кости по отношению к локтевой, дорсальный, поддающийся редрессации подвывих дистального конца локтевой кости (деформация Маделунга).
- к) Пикнодизостоз: короткие концевые фаланги, большой мозговой череп, большие роднички (которые могут быть открытыми до зрелого возраста), спонтанные переломы.
- л) Карликовость с микромелией: гипоплазированные локтевая и малоберцовая кости, а также нижняя челюсть.
- м) Карликовость с микромелией: характерная «треугольная» деформация большеберцовой кости.
- н) Псевдогипопаратиреоз: ожирение, обращающее на себя внимание круглое лицо, укороченные пальцы рук, изменения метаболизма.
- о) Псевдопсевдогипопаратиреоз.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник
Седых А.В. 1
1Тюменский государственный медицинский университет Министерства здравоохранения Российской федерации, Тюмень, Россия
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Введение
Актуальность. Низкорослость (нанизм) – достаточно часто встречающийся симптом различных наследственных, врождённых и приобретённых заболеваний детского возраста. Одним из примеров таких заболеваний является синдром Секкеля (сочетание карликовости с «птицеголовостью», микроцефалическая примордиальная карликовость I типа), в основе которого лежат мутации различных генов.
Цель: изучить развитие болезниСеккеля, ее этиологию, частоту встречаемости и
патогенез, а также лечение.
Задачи:
1) Проанализировать этиологию и встречаемость болезни
2) Изучить патогенез и клиническую картину болезни
3) Рассмотреть лечение болезни
Этиология и встречаемость
Впервые синдром описали Тревор Манн и Александр Рассел в 1959 году. Гельмут Секкель в 1960 году опубликовал статью, в которой привёл результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления, а термины «птицеголовая карликовость» и «наноцефалия» принадлежат Рудольфу Вирхову.
Частота встречаемости в популяции составляет 1:10 000 случаев. Согласно генетическим данным, заболевание наследуется по аутосомно-рецессивному типу.(Описано 9 форм заболевания)
Развитие синдрома Секкеля 1 связывают с мутацией в гене ATR, локализованном на хромосоме 3q23. Ген является важнейшим регулятором геномной целостности, контролирует и координирует ДНК-репликацию. Поломки в гене ATR приводят к нарушению клеточного цикла и появлению нестабильности хрупких хромосомных участков, что связывают с развитием фенотипа синдрома Секкеля 1. Мутации в гене АТR отвечают также за развитие атаксии-телеангиэктазии и семейных форм рака.
Дети с синдромом Секкеля рождаются с выраженной внутриутробной гипоплазией: средняя масса тела доношенных новорождённых составляет 1600-1700 г, длина тела – 40-45 см. В последующие годы сохраняется пропорциональная низкорослость, чаще значительная, масса тела соответствует росту.
Патогенез
Наиболее характерными внешними признаками заболевания являются черепно-лицевые дизморфии, давшие основание для термина «птицеголовость»: микроцефалия с преждевременным закрытием черепных швов (краниосиностоз); задержка увеличения окружности головы, в поло- вине случаев соответствующая общей задержке роста, а в половине – даже превосходящая ее; скошенный лоб; выступающий загнутый («клювовидный») нос с утолщенным кончиком; микро- и ретрогения; низкорасположенные деформированные ушные раковины; относительно большие глаза с антимонголоидным разрезом; редкие волосы . Отмечаются также страбизм, аномалии зубов и ротовой полости (узкое высокое нёбо, его расщелины, частичная адонтия, гипоплазия эмали). Синдром может сопровождаться появлением гипопигментированных пятен на различных участках тела – наиболее часто они наблюдаются в области подмышечных впадин и на шее.
Диагностика
Диагностика заболевания основывается на клинических проявлениях. В пренатальной диагностике основную роль играет ультразвуковое исследование, которое позволяет выявить гипоплазию плода, микроцефалию, аномалии развития конечностей, черепно-лицевые аномалии. При данном синдроме возможно перенашивание беременности.
Лечение
При синдроме Секкеля лечение симптоматическое. Применяются препараты рекомбинантного гормона роста (рГР), препараты, улучшающие обменные процессы, ноотропы . Несмотря на выраженную задержку психического развития, раннее двигательное развитие может соответствовать возрасту. Больные обычно дружелюбны и общительны, но в то же время невнимательны и неусидчивы. Известно об одном больном синдромом Секкеля, дожившем до 75 лет.
Список литературы
Роберт Л. Ньюссбаум, Родерик Р. Мак-Иннес, Хантингтон Ф. Виллард «Медицинская генетика» (с.485-486)
https://simptomy-lechenie.net/ — Синдром Секкеля
Логачев М.Ф. Задержка внутриутробного развития и постнатальная низкорослость у детей: (Этиология, патогенез, клиническая картина, диагноз): дис. . д-ра мед. наук / Логачев М.Ф. М., 2000. — 289 с.
Источник
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) — редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птице- головость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефа- лия» принадлежат Н.ЛгсГюж Н.Зеске! в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Частота встречаемости в популяции составляет 1 : 10 000 [1Шарреп, 1993].
Гдиетические данные и патогенез. Заболевание наследуется по аутосомно-ре- цессивному типу. Описаны случаи синдрома Секкеля у сибсов обоего пола; имеются сведения о повышенной частоте кровного родства родителей; ни у одного из родителей не отмечалось клинических проявлений данного заболевания.
Синдром Секкепя является генетически гетерогенной патологией В настоящее время выделено два типа этого заболевания. При I типе ген локализован на длинном плече хромосомы 3 в регионе Зд22.1- ц24 [75]. Ген синдрома Секкеля II типа картирован на хромосоме 18 в локусе 18р11.31-д11.2 [76]. Генетическую гетерогенность этого заболевания подтверждают результаты недавно проведенного молекулярно-генетического исследования у нескольких больных с характерными фенотипическими проявлениями, у которых не было выявлено мутаций в локусах синдрома Секкеля I и II типов [77]
Патогенез до настоящего времени остается неизученным.
У некоторых больных с клиническими признаками синдрома Секкеля при цитогенетическом исследовании выявляется хромосомная нестабильность в виде увеличения частоты разрывов и сестринских хро- матидных обменов. У этих пробандов имеется склонность к развитию анемии типа Фанкони, панцитопении, острого мие- лобластного лейкоза. У некоторых больных с гематологическими нарушениями имеется повышение чувствительности к митоми- цину С.
Пороки и аномалии развития центральной нервной системы, выявленные у нескольких детей на основании результатов аутопсии или МРТ головного мозга, позволили высказать некоторым исследователям предположение о том, что при этом заболевании может иметь место нарушение нейрональной миграции [78].
Клинические проявления. Дети рождаются с выраженной внутриутробной гипоплазией’ средняя масса тела доношен
ных новорожденных 1 600-1 700 г, длина тела — 40-45 см В дальнейшем сохраняет ся значительное пропорциональное отста вание в физическом развитии
Характерны черепно-лицевые аномалии микроцефалия, узкое лицо, маленькая нижняя челюсть, выступающий клювовидный нос и большие глаза Перечисленные признаки послужили основанием для термина «птицеголовость» (рис 7 1 7 на цветной вкладке) У большинства больных отмечаются низкорасположенные дефор мированные ушные раковины, часто увеличенные в размерах, редкие волосы Могут наблюдаться антимонголоидный разрез глаз, узкое высокое небо, расщелина неба, ретрогнатия, частичная адентия, гипоплазия эмали зубов В трети случаев выявляется краниосиностоз
Из скелетных аномалий часто отмечаются кифоз, сколиоз, вывихи крупных суставов, плоскостопие, косолапость, сан далевидная щель У отдельных больных также выявляются брахидактилия, клино- дактилия мизинцев, гипоплазия больших пальцев кистей, гипоплазия или аплазия нижнего ребра, отсутствие эпифизов некоторых фаланг, гипоплазия проксимальной части лучевой кости
Подавляющее большинство больных с синдромом Секкеля умственно отсталые Задержка психомоторного развития становится заметной спустя несколько месяцев после рождения Степень снижения интеллекта вариабельна, но чаще всего наблюдаются умеренная или глубокая умственная отсталость У отдельных больных отмечаются судороги
Часто встречается патология органов зрения и слуха Увеличение диаметра радужки у детей с этим заболеванием создает впечатление больших глаз Описаны катаракта, колобома радужки, косоглазие У некоторых больных выявляется сенсо- невральная тугоухость
Пороки внутренних органов нетипичны для синдрома Секкеля, однако у отдельных больных описаны сердечно-сосудистые аномалии, аноректальные дефекты У мальчиков иногда выявляются крипторхизм, гипо плазия наружных половых органов, у дево чек — гипертрофия клитора Имеются сведения об эндокринных нарушениях у некото рых пробандов
Клинические отличия синдромов Секкеля I и II типов представлены в табл 7 14 Диагностика основывается на клиниче ских проявлениях
Детям с этим заболеванием необходимо проводить цитогенетическое исследование для выявления повышенного уровня хромосомных перестроек, так как при наличии признаков хромосомной нестабильности у больных имеется высокий риск развития тяжелых гематологических нарушений При МРТ-исследовании головного мозга обнаруживаются такие аномалии, как аге незия мозолистого тела, дизгенезия церебральной коры, пахигирия
Дифференциальная диагностика проводится с врожденными формами нанизма с микроцефалией и, в первую очередь, с примордиальной карликовостью с остеодисплазией I и II типов и синдромами Дубовица, Корнелии де Ланге, а также с монреальским типом карликовости с «птичьей головой», — для которого характерно отсут ствие пренатальной гипоплазии и наличие признаков преждевременного старения в виде раннего поседения, облысения и морщинистой кожи
Лечение симптоматическое Возможно применение ноотропных препаратов для стимуляции психического развития
Специфической профилактики синдрома Секкеля не существует Важное значение имеют пренатальная диагностика и выявле ние при ультразвуковом исследовании плода выраженной задержки внутриутробного развития в сочетании с микроцефалией
1 Козлова С И. Демикова Н С Семанова Е Блинникова ОЕ Наследственные синдромы и медико-генетическое консультирование М Практика, 410
2 Рай(|е1й С Л РаПтд(оп М №, Зкпрзоп N Е ТИе ЙиЬт51ет-ТауЬ| зупйготе Агсй 015 С1и1й1968
43 94-101
3 (.асотЬе й, 8аига Й, Тате I, ВаНт з Соп(|Г- та(юп о{ азз1дптеп( о? а 1осиз (ог ВиЬтз- (ет-ТауЫ зупйготе депе (о16р13 3 Ат ^ Мей 6епе( 1992,44 126-8
4 Ш1егз(ет В, Апйегзоп СЕ, Нау В, е( а1 ЗиЬтюгозсорю йе1е(юпз а( 16р13 3 т ВиЬт- з(ет-ТауЬ| зупйготе (гедиепсу апй сНшса! тат(е51а(юп5 т а ИоПН Атепсап рори1а(юп ^ Мей 6епе( 1997 34 203-6
5 1та1гит1 К, Кигок! V КиЬт51ет-ТауЬ| зупйготе №1(11 йе поуо гесфгоса! (гапз1оса1юп ?(2,16) (р13 3,р13 3) Ат ^ Мей 6епе( 1991, 38 636-9
6 Тоттегир N , уап йег Надеп С В, Не1Ьегд А Теп(а(|уе азз1дптеп( о( а 1осиз (ог ЙиЬт- з(ет-ТауЬ1 зупйготе (о 16р13 3 Ьу а йе поуо георгоса) (гапз1оса(юп, ?(7,16)(Ноодепгаас! С С , Коеккоек В, АкЬтапоуа А е» а1 Тагде(еэто заболевание, соответственно, в 1963 и 1964 годах.
Частота патологии — 1 :10 000-1 :15 000 новорожденных [1].
Генетические данные и патогенез. Тип наследования синдрома неоднозначен и довольно сложен. В 15% семейных случаев характер передачи заболевания определяется как аутосомно-доминантный. У большинства больных (примерно 80%) патология возникает спорадически.
Синдром Беквита-Видемана обусловлен различными повреждениями кластера генов, расположенных в области короткого плеча хромосомы 11, в сегменте 15.5 [2].
Большую роль в нормальном функционировании генов этого локуса играет геномный импринтинг. Импринтинг — это различное действие генов в зависимости от их родительского (отцовского или материнского) происхождения. В настоящее время нарушение импринтинга считается основной причиной развития синдрома Беквита-Видемана.
У некоторых больных заболевание обусловлено хромосомной патологией — деле- цией, дупликацией или транслокацией с вовлечением локуса 15.5 короткого плеча хромосомы 11. На долю таких хромосомных аномалий приходится, примерно, 2% [3].
Клинические проявления. Заболевание формируется внутриутробно. Обращают на себя внимание высокие антропометрические параметры новорожденного: в среднем, длина тела составляет 52,6 см, масса — 4 кг. Высокие показатели физического развития сохраняются и в дальнейшем. Характерны также макро- глоссия и грыжа пупочного канатика (ом- фалоцеле). Типичными признаками синдрома являются горизонтальные насечки на мочках ушей, микро- или макроцефалия, выступающий затылок, экзофтальм (вследствие относительной гипоплазии орбит), пупочная и паховая грыжи, клито- ромегалия, висцеромегалия (печени, почек, поджелудочной железы, иногда сердца). Нередко встречаются пороки развития внутренних органов: двурогая матка, диафрагмальная грыжа, добавочная селезенка, врожденные пороки сердца — чаще септальные дефекты, легких (неправильная дифференцировка легочной ткани на доли), кишечника (незавершенный поворот кишечника).
Психическое развитие больных варьирует: описаны пациенты как с нормальным интеллектом, так и с умственной отсталостью различной степени выраженности (рис. 7.2.1 на цветной вкладке).
Диагностика и дифференциальная диагностика. При обследовании новорожденных с синдромом Беквита-Видемана, как правило, выявляются полицитемия и гипогликемия, приводящие к развитию тяжелой неврологической симптоматики и, нередко, к летальному исходу.
У детей старше 7-10 лет часто диагностируют гиперлипидемию, гиперхолесте- ринемию, гипокальциемию [4].
Для детей с синдромом Беквита-Видемана характерно повышенное содержание в сыворотке крови соматомедина С, играющее, вероятно, ведущую роль в реализации высоких антропометрических параме-
трое больных [5] Высокий уровень сома- томедина С в сыворотке крови детей с синдромом Беквита-Видемана обусловлен избыточной продукцией гена инсули ноподобного фактора роста-2 (ген ЮР2), возникающей в результате потери геномного импринтинга (диаллельная экспрессия гена 1(3 Р2)
При синдроме Беквита-Видемана нередко регистрируются иммунодефицит- ные состояния Заболевание относится к группе риска развития злокачественных новообразований Одной из возможных причин формирования опухолей может быть снижение адаптивного ответа, выявляемое у больных с синдромом Беквита-Видемана [5] Суть его заключается в том, что предварительная обработка лимфоцитов малыми (неповреждающими) дозами мутагена с чувствительностью клеток к воздействию больших (повреждающих) доз этого же самого или других мутагенов Адаптивный ответ оценивается по процентному содержанию двуните- вой ДНК У здоровых детей адаптивный ответ, как правило, нормальный и равен 100% У лиц с синдромом Беквита-Виде- мана процент двунитевой ДНК после двойной обработки лимфоцитов у-лучам и в возрастающей дозировке составяет не выше 75%, что свидетельствует о полном отсутствии у больных адаптивного ответа по у-типу
Рентгенологические методы исследования позволяют выявить опережение костного возраста, расширение метафизов длинных трубчатых костей и кольцевидное сужение диафизов
Патоморфологические изменения при патолого-анатомическом анализе нередко обнаруживают гиперплазию клеток островков Лангерганса, нефрогенную бласто- му, резкое увеличение клеток и ядер в надпочечниках
Дифференциальная диагностика проводится с заболеваниями, которым свойственны высокие антропометрические параметры и снижение интеллекта К таким болезням относятся синдромы Сотоса, Симпсона-Голаби-Бехмеля, Вивера, Маршалла-Смита, аномалии половых хромосом (синдромы Клайнфельтера — 47, XXV и 47, ХУУ) и омфалоцеле
Лечение синдрома Беквита-Видемана носит симптоматический характер и направлено, в основном, на стимуляцию нер- вно-психического развития и иммунного статуса больных
С целью профилактики развития злокачественных новообразований необходимо оберегать больных от неблагоприятных воздействий внешней среды строго запрещены профессии, связанные с радиацией и химическими веществами, противопоказано проживание в районах с повышенным радиационным фоном и высокой инсоляцией Показан также строгий контроль за назначением цитостати- ческих препаратов и ионизирующего облучения
7.2.2.
Еще по теме Синдром Секкеля:
- Секкеля синдром
- СИНДРОМ ДИССЕМИНИРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ КРОВИ (ДВС-СИНДРОМ, ТРОМБОГЕМОРРАГИЧЕСКИЙ СИНДРОМ)
- Глава 3СИСТЕМА ГЕМОСТАЗА ПРИ ГЕСТОЗЕ. ГИПЕРКОАГУЛЯЦИОННЫЙ СИНДРОМ. ДВС-СИНДРОМ
- СИНДРОМ ПОЗВОНОЧНОГО НЕРВА 13АДНИЙ ШЕЙНЫЙ СИМПАТИЧЕСКИЙ СИНДРОМ)
- СИНДРОМ ШТЕЙНА—ЛЕВЕНТ АЛЯ (СИНДРОМ ОВАРИАЛЬНОЙ ГИПЕРАНДРОГЕНИИ НЕОПУХОЛЕВОГО ГЕНЕЗА)
- СИНДРОМ НАРУШЕНИЯ КРОВООБРАЩЕНИЯ В КОНУСЕ И ЭПИКОНУСЕ СПИННОГО МОЗГА (СИНДРОМ АРТЕРИИ ЭПИКОНУСА1
- СИНДРОМ СЕГМЕНТАРНО-ДИССОЦИИРОВАННОГО РАССТРОЙСТВА ЧУВСТВИТЕЛЬНОСТИ С АМИОТРОФИЕЙ ПЛЕЧА (СИРИНГОМИЕЛИЧЕСКИЙ СИНДРОМ)
- Синдром амблиопии, болевой нейропатии и орогенитального дерматита (синдром 81гаскап) _
- Менингеапьный синдром. Повышение внутричерепного давления (гипертензионный синдром). Гидроцефалия
- Церебральный сольтеряющий синдром и синдром гиперсекреции АДГ после субарахноидального кровоизлияни
- Синдром полигландулярной недостаточности (синдром Шмидта) (см. гл. 324 и 325)
- Атаксия-телеангиэктазия (синонимы: синдром Луи-Бар, синдром Бодера-Седжвика).
- Атаксия-телеангиэктазия (синонимы: синдром Луи-Бар, синдром Бодера-Седжвика).
Источник