Прогноз при синдроме беквита видемана
Синдром Беквита-Видемана. Диагностика и прогноз синдрома Беквита-Видемана
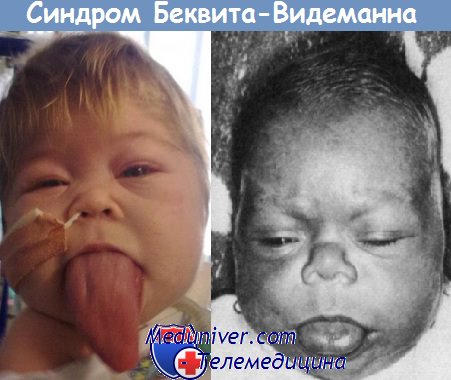
Синдром Беквита-Видемана (Beckwith-Wiedemann) впервые был описан J.В. Beckwith в 1963 году и H.R. Wiedemann в 1964 году. Этот синдром характеризуется классической триадой, включающей макросомию, омфалоцеле и макроглоссию.
Синонимы. Синдром экзомфалии — макроглоссии — гигантизма.
Распространенность. Частота встречаемости оценивается на уровне 0,72 на 10 000 родов. В литературе описано более 500 клинических наблюдений этого заболевания.
Этиология. В большинстве случаев синдром Беквита-Видемана (Beckwith-Wiedemann) возникает спорадически и имеет аутосомно-рецессивный тип наследования с неполной пенетрантностью и вариабельной экспрессивностью. Предполагается, что данное заболевание может возникать вследствие перестроек вовлекающих регион короткого плеча хромосомы 11 р15.
Выявление макросомии, омфалоцеле и макроглоссии в сочетании с нормальным кариотипом позволяет диагностировать синдром Беквита-Видермана (Beckwith-Wiedemann). Другие признаки, которые обнаруживаются с переменной частотой, включают нефромегалию, гепатомегалию, многоводие, складки мочек уха, диафрагмальную грыжу и пороки сердца. В 30-50% случаев встречается гиперплазия клеток поджелудочной железы, что вызывает гиперинсулинизм и неонатальную гипогликемию, манифестирующую на второй или третий день жизни. Небольшая складка на мочке уха является довольно типичным признаком этого синдрома.
При цитогенетических исследованиях могут быть выявлены структурные хромосомные аномалии, включая изодисомию части короткого плеча хромосомы 11р15.5, изодисомию всего короткого плеча 11q и однородительскую дисомию этой хромосомы.
Поскольку при синдроме Дауна (Down) также наблюдается макроглоссия, этот синдром должен быть исключен путем проведения хромосомного анализа. Причиной макросомии обычно является диабетическая фетопатия, поэтому вероятность этого состояния тоже должна учитываться при проведении дифференциального диагноза. Нормальный уровень глюкозы в крови матери помогает его исключению. При синдроме Зельвегера (Zellweger) может отмечаться увеличение печени и почек, которое пренатально диагностируется путем измерения концентрации жирных кислот и активности соответствующих ферментов.
Некоррегированная неонатальная гипогликемия является серьезным осложнением, что может повлечь за собой дальнейшие церебральные дисфункции, такие как судороги, умственная отсталость, от слабой до умеренно выраженной, или в более тяжелых случаях даже стать причиной неонатальной гибели. Макроглоссия может вызывать различные патологические состояния, начиная от затруднений при вскармливании до обструкции дыхательных путей и гибели. Отдаленные осложнения включают высокий риск развития опухолей органов брюшной полости, в особенности опухоли Вильмса (Wilm), гепатобластомы, нейробластомы и злокачественной опухоли коры надпочечника.
Уровень неонатальной смертности составляет примерно 21% и в основном обусловлен застойной сердечной недостаточностью. Для выживших прогноз в целом бывает благоприятным и зависит от тяжести сочетанных аномалий и наличия отдаленных осложнений.
До наступления периода жизнеспособности плода может быть предложено прерывание беременности. В более поздние сроки проводится эхографическая оценка развития плода в динамике. В случаях подозрения на макросомию может быть предложено кесарево сечение из-за риска развития дискоординации родовой деятельности. Целесообразно родоразрешение в специализированных пренатальных медицинских центрах для проведения раннего хирургического лечения дефектов передней брюшной стенки и коррекции гипогликемии. Рекомендуется эхографическое обследование ребенка каждые три месяца в течение первых 6 лет жизни для выявления опухолей органов брюшной полости.
— Вернуться в оглавление раздела «Акушерство.»
Оглавление темы «Врожденные пороки развития плода»:
1. Особенности диагностики фетальных синдромов. Ахондрогенез и его частота
2. Генетические нарушения при ахондрогенезе. Ахондроплазии у плода
3. Дифференциальный диагноз ахондроплазии у плода. Синдром акрофациального дизостоза — синдром Нагера
4. Акромезомелическая дисплазия. Синдром Эйкарди
5. Врожденный СПИД. Фетальная ВИЧ-инфекция
6. Синдром амниотических перетяжек. Диагностика и прогноз при синдроме амниотических перетяжек
7. Синдром Апера у плода. Диагностика и прогноз при синдроме Апера
8. Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
9. Асфиксическая дисплазия грудной клетки. Синдром асплении и полисплении
10. Синдром Беквита-Видемана. Диагностика и прогноз синдрома Беквита-Видемана
Источник
Синдром Беквита — Видемана (англ. BWS — Beckwith-Wiedemann-Syndrom, EMG — Exomphalos-Makroglossie-Gigantismus, также синдром «гигантизма с пуповинной грыжей») — одна из редких генетических аномалий, связана с превышением норм роста плода при беременности и с несогласованным развитием различных отделов организма.
Типичными признаками синдрома являются увеличенные размер и вес плода (новорождённого ребёнка), причем происходят не только асимметричный рост организма и внешние отклонения в размерах тела, но и непропорционально большой размер некоторых внутренних органов: печени, селезёнки, почек и языка.[1]
Особенности синдрома[править | править код]
На наличие синдрома может указывать наличие одного или более из следующих факторов:
- Пренатальное и постнатальное опережение роста (таковым обычно считают рос и вес выше 97 процентили)
- Макроглоссия
- Омфалоцеле (грыжа пупочного канатика, пуповинная грыжа, эмбриональная грыжа — грыжевой мешок, сформированный брюшиной, выходит из брюшной полости через пупочное отверстие (пупок))
- Висцеромегалия
- Эмбриональные опухоли (такие как гепатобластома, нефробластома, рабдомиосаркома) в детском возрасте
- Гемигиперплазия (асимметричный рост одной или нескольких частей тела)
- Почечные аномалии
- Адренокортикальная цитомегалия
- Неонатальная гипогликемия
Частые симптомы[править | править код]
- Лёгкая или умеренная умственная отсталость (с неуточнённой частотой).
- Рост:
- Макросомия с увеличением мышечной массы и подкожного жирового слоя;
- ускоренное созревание костной ткани;
- широкие метафизы и узкие диафизы длинных трубчатых костей;
- слабо заметный переход проксимального метафиза плечевой кости в диафиз.
- Голова:
- макроглоссия;
- экзофтальм — смещение глазного яблока вперёд (выпученные глаза) с относительным недоразвитием подглазничного края;
- пламенеющий невус на лбу и веках;
- выступающий лобный шов;
- широкие роднички;
- выступающий затылок;
- неправильный прикус из-за нижней прогнатии (выступающая вперед нижняя челюсть) и верхней микрогнатии (врождённое недоразвитие челюстной кости);
- насечки на мочках ушей и задней поверхности завитков.
- Внутренние органы:
- нефромегалия (увеличение размеров одной или обеих почек) с дисплазией (неправильным развитием) мозгового вещества почек;
- панкретомегалия (увеличение размеров поджелудочной железы) с гиперплазией (размножением клеток и образованием новых тканевых структур) островков поджелудочной железы;
- гипертрофия (увеличение объёма и массы) клеток коры надпочечников у плода (постоянный признак);
- гиперплазия интерстициальных клеток половых желез (межуточных клеток, расположенные в строме яичников и между канальцами семенников у млекопитающих; участвуют в выработке половых гормонов: в семенниках — андрогенов, в яичниках — эстрогенов[2]) и амфифильных клеток аденогипофиза.
- Прочее:
- эритроцитоз (увеличение количества эритроцитов) у новорождённых;
- гипогликемия в раннем детском возрасте (30-50 %);
- грыжа пупочного канатика и другие аномалии пуповины; расхождение прямых мышц живота;
- релаксация диафрагмы;
- крипторхизм (неопущение яичка в мошонку);
- врождённые пороки сердца, в том числе изолированная кардиомегалия.[3]
Этиология и встречаемость болезни[править | править код]
Синдром Беквитта-Видемана — панэтнический (не связанный с принадлежностью к какой-либо национальности) синдром. Возникает спорадически, иногда наследуется по аутосомно-доминантному типу. Встречается приблизительно у одного из 13 700 родившихся живыми детей[1].
Синдром Беквитта-Видемана вызван нарушением баланса экспрессии импринтированных генов в регионе р15 хромосомы 11[1].
Среди генов, расположенных к этом регионе, находятся кодирующие белки гены CDKN1C и IGF2:
- CDKN1C кодирует супрессор клеточного цикла, ограничивающий деление и рост клеток;
- IGF2 кодирует инсулиноподобный фактор роста, стимулирующий рост.
Также в этом регион расположены транскрибируемые, но не транслируемые KCNQOT1 и Н19. Их транскрипция подавляет экспрессию отцовской копии CDKN1С и материнской копии IGF2 соответственно: в норме эти гены импринтированы и экспрессируются только из отцовского (IGF2 и KCNQOT1) или только материнского аллеля (HI9 и CDKN1C)[1].
Ссылки[править | править код]
- Безуглаяя А. А. Синдром Беквита — Видемана
- О синдроме на сайте medaboutme.ru
Примечания[править | править код]
Источник
Синдром Беквита – Видемана — нарушение физического развития человека. Заболевание может проявляться яркими симптомами или протекать незаметно. Чаще обращают внимание на внешние признаки, которые хорошо видны визуально: когда ребенок при рождении имеет массу тела свыше четырех килограммов, рост — от пятидесяти шести сантиметров. При синдроме различные части тела непропорциональны и даже асимметричны: часто увеличен язык, большие уши, слишком пышные щеки.
Онлайн консультация по заболеванию «Синдром Беквита – Видемана».
Задайте бесплатно вопрос специалистам: Хирург.
Есть видимые дефекты брюшины, например грыжа. Детки склонны к новообразованиям, врожденным порокам сердца. Диагностируются увеличенные внутренние органы и низкий уровень сахара в крови (гиперинсулинизм), что может вызывать судороги. Если патология слишком тяжелая, возможен летальный исход.
Часто люди даже не знают о такой патологии, как синдром Беквита-Видемана, но болезнь диагностируют нередко: согласно статистическим данным, на каждые четырнадцать тысяч новорожденных рождается один ребенок с патологией.
Заболевание наследственного характера, развивается из-за изменений в одиннадцатой хромосоме. Выявить патологию в пубертатном периоде нельзя. В девяноста процентах случаев в семейном анамнезе родителей генетических отклонений не было. Остальные двадцать процентов передают болезнь по наследству.
Появиться синдром Беквита-Видемана может из-за таких причин:
- генетический фактор;
- спонтанная мутация.
В случае наследования передача происходит по аутосомно-доминантному типу: патология передается как мальчикам, так и девочкам. Симптомы заболевания не всегда будут проявляться у одного из родителей. Специалисты объясняют это как мозаичную форму, когда половина клеток с нормальным генотипом, а половина — изменены.
Бывает и спонтанное появление синдрома по невыясненным пока причинам, которые оказывают влияние на клетки зародыша еще в самом начале беременности.

Признаки синдрома Беквита-Видемана
Патология имеет как внешние, так и внутренние признаки, указывающие на ее развитие:
- ребенок начинает быстро расти с раннего детства — темп роста начинает замедляться после восьми лет;
- в некоторых случаях аномальное развитие частей тела происходит только с одной стороны, поэтому возникает асимметричность внешнего вида;
- у некоторых детей слишком увеличивается язык, что вызывает трудности при глотании и дыхании;
- сильно увеличиваются органы брюшной полости;
- наблюдаются увеличенные ушные раковины;
- аномальное развитие почек;
- низкий уровень сахара в крови.
У детей повышен риск развития онкологии. Новообразованиям подвержены почки, могут возникнуть гепатобластомы и опухоль Вильмса.
При обращении будущих родителей к доктору будет проводиться генетическое тестирование, которое покажет возникшую мутацию. Это сложная процедура, помогающая диагностировать синдром в 80 % случаев.
Консультацию должен провести врач-генетик, который подскажет, какое обследование необходимо пройти, ведь процедура назначается индивидуально.
Выявить синдром Беквита-Видемана можно и у беременной женщины. Признаки патологии проявляются как осложнения при беременности:
- большое количество околоплодных вод;
- слишком высокий риск преждевременной родовой деятельности.
Выявить патологии можно во время проведения УЗИ.
Здоровье ребенка необходимо постоянно контролировать — если понадобится, оказать ему врачебную помощь.
- в индивидуальном порядке необходимо следить за уровнем сахара в крови;
- в течение жизни больные люди должны внимательно следить за здоровьем, потому что высок риск возникновения новообразований.
Необходимо отметить, что взрослые люди, которым в раннем детстве ставили такой диагноз, вполне нормально живут, не испытывая дискомфорта.
Если у новорожденного слишком увеличен язык, назначают оперативное вмешательство. В ходе проведения операции выполняют коррекцию формы языка. Избежать хирургического вмешательства не удастся, потому что дефект будет создавать проблемы во время кормления ребенка и при дыхании. В будущем речь будет искажена.
Проблему с увеличенными внутренними органами и пупочной грыжей решают с помощью хирургического вмешательства.
Консультацию проводят иммунолог и ортопед.
Таким детям очень нужна забота родителей. Ребенок не должен находиться на сквозняке, переохлаждаться. К тому же его необходимо оберегать от инфекционных заболеваний, ведь у больных детей часто низкий иммунитет, когда даже обычное респираторное заболевание может оказаться большой проблемой. Родители даже при небольших проблемах со здоровьем должны обращаться за помощью к специалисту.
В качестве профилактики еще до планирования беременности можно пройти специальные тесты, которые помогут обнаружить изменения хромосом у будущих родителей. Об этом задумываются редко, узнавая об отклонении уже после рождения ребенка с таким диагнозом.
Заболевание чаще передается по наследству, поэтому если в семье кто-то болен, вероятность рождения ребенка с патологией составляет 55 %.
На сегодняшний день, к сожалению, нет медикаментозного лечения синдрома. Если пара, где один из родителей или оба больны, хочет завести детей, единственным методом рождения здорового ребенка будет искусственное оплодотворение.
Прогноз для жизни вполне благоприятен, если вовремя выявить болезнь и оказать ребенку медицинскую помощь. За новорожденным должны хорошо ухаживать родители. Спустя некоторое время темп роста приходит в норму, а пропорции тела восстанавливаются.
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знания
Читать нас на Яндекс.Дзен
Заболевания со схожими симптомами:
Вывих нижней челюсти — патологическое состояние, суть которого заключается в смещении суставной головки со своей анатомической позиции, т. е. она соскальзывает на передний скат суставного бугорка височной кости. Такие изменения приводят к стойкому нарушению функционирования ВНЧС. Частота распространенности варьируется от 1,5 до 5,5 % среди всех вывихов.
…
Синдром Клиппеля-Фейля — врожденное аномальное явление шейного отдела позвоночника, которое характеризуется сращением и уменьшенным количеством позвонков. Основным ярко выраженным признаком патологии выступает малоподвижная и короткая шея.
…
Гипертрофия миндалин – патологический процесс, при котором происходит увеличение лимфоидных узлов, которые расположены между передними и задними небными дужками. Клиническая картина на раннем этапе развития отсутствует, а в целом симптомы неспецифические.
…
Лёгочная недостаточность – состояние, характеризующееся неспособностью лёгочной системы поддерживать нормальный газовый состав крови, или же он стабилизируется за счёт сильного перенапряжения компенсаторных механизмов аппарата внешнего дыхания. Основа данного патологического процесса – нарушение газообмена в лёгочной системе. Из-за этого в тело человека не поступает требуемый объем кислорода, а уровень углекислого газа постоянно возрастает. Все это становится причиной кислородного голодания органов.
…
Омфалоцеле (совпадающих симптомов: 2 из 13)
Омфалоцеле (пуповинная или эмбриональная грыжа, грыжа пупочного канатика) — врожденная патология, когда происходит выпадение органов брюшной полости через среднюю линию в основании пупка. Встречается патология у 1–2 младенцев на 10 тысяч новорожденных.
…
Источник
Синдром Беквита – Видемана
Синдром Беквита – Видемана (СБВ, BWS, EMG, синдром «гигантизма с пуповинной
грыжей», МКБ 10 – Q87.3) — редкое наследственное заболевание, характеризующееся
триадой признаков: омфалоцеле (центральный дефект передней брюшной стенки в
области пупочного кольца, через который из брюшной полости выпячиваются внутренние
органы, покрытые грыжевым мешком), макроглоссией (аномально большой язык) и
макросомией (крупные размеры тела).
Развитие синдрома Беквита – Видемана связано с нарушением баланса экспрессии
импринтированных генов в регионе 11p15.5 (KCNQOT1, Н19, CDKN1C и IGF2).
Синдром Беквита – Видемана впервые описали в 1969 г. патолог из США Дж. Б.
Беквит (J. B. Beckwith) [1] и германский педиатр Х.-Р. Видеман (H.-R. Wiedemann) [2].
Одно из названий синдрома Беквита – Видемана — EMG — образовано из первых букв
триады основных клинических симптомов: Exomphalos, Makroglossie, Gigantismus.
В 1999 г. был картирован ген KCNQOT1, мутации в котором связаны с 50 %
спорадических случаев синдрома Беквита – Видемана [8], а в 1995 г. был картирован
второй по значимости для развития заболевания ген — CDKN1C [7].
Распространенность и тип наследования
Синдром Беквита – Видемана встречается во всем мире со средней частотой 1
случай на 13 700 новорожденных. Болезни в равной мере подвержены и мальчики, и
девочки [3].
В подавляющем большинстве случаев синдром Беквита – Видемана является
спорадическим, т. е. генетическая мутация возникает впервые в семье. При семейной
форме заболевания синдром Беквита – Видемана имеет аутосомно-доминантный тип
наследования: достаточно получить от родителей хотя бы одну копию «дефектного» гена,
чтобы проявилась патология [4].
Синдром Беквита – Видемана дебютирует на первом году жизни. Родители
обращаются к врачу с жалобами на чрезмерный рост и вес ребенка, большой язык,
асимметрию тела, дефект передней брюшной стенки: омфалоцеле или пупочную грыжу.
Первичную диагностику проводит педиатр. При подозрении на синдром Беквита –
Видемана пациенту назначают анализ крови на глюкозу, инсулин, кальций и креатинин.
Также проводят КТ органов грудной клетки, МРТ и УЗИ брюшной полости,
рентгенографию грудной клетки, ЭХО КГ и ЭКГ. Для постановки клинического диагноза
СБВ требуется наличие по крайней мере трёх «больших» признаков или двух «больших»
и одного «малого» признака заболевания.
К «большим» признакам относятся:
- дефект передней брюшной стенки: омфалоцеле или пупочная грыжа,
- макроглоссия,
- макросомия,
- передние складки мочки уха и/или задние спиральные ямки (двусторонние или односторонние),
- увеличенные размеры (висцеромегалия): печени, почек, селезенки, поджелудочной железы и надпочечников,
- эмбриональное опухоли в детском возрасте,
- асимметричный рост одной или нескольких частей тела — гемигиперплазия,
- цитомегалия коры надпочечников плода, как правило диффузная и двусторонняя,
- аномалии почек, в том числе медуллярная дисплазия с последующим развитием медуллярной губчатой почки,
- отягощенность семьи по синдрому Беквита – Видемана,
- расщелина нёба.
К «малым» признакам относятся:
- многоводие, большая плацента и/или толстая пуповина при беременности,
- преждевременные роды,
- неонатальная гипогликемия,
- пламенеющий невус (или «винные пятна») на теле,
- кардиомегалия / структурные аномалии сердца / кардиомиопатия,
- характерное лицо,
- диастаз прямых мышц живота,
- опережение костного возраста [3].
Для подтверждения диагноза синдрома Беквита – Видемана проводят
молекулярно-генетический анализ генов CDKN1C, H19, KCNQ1OT1 и IGF2 [3].
В неонатальный период пациенты с синдромом Беквита – Видемана подвержены
риску гипогликемии, которая приводит к поражению головного мозга. Поэтому таким
больным необходимо введение глюкозы.
Хирургическая помощь при синдроме Беквита – Видемана нужна, если у пациента
наблюдается омфалоцеле, требующее оперативного лечения, или различные опухоли. При
выраженной гемифациальной гиперплазии показана пластическая коррекция лица [3].
Прогноз при синдроме Беквита – Видемана напрямую зависит от сроков начала и
адекватности специализированного лечения. При постоянной корректировке стойкой
гипогликемии прогноз в целом благоприятный [5].
Развитие синдрома Беквита – Видемана связано с нарушением баланса экспрессии
импринтированных генов в регионе 11p15.5. В этом регионе расположены
транскрибируемые, но не транслируемые гены KCNQOT1 и Н19 и кодирующие белки
гены CDKN1C и IGF2. В норме эти гены импринтированы и экспрессируются только из
отцовского (IGF2 и KCNQOT1) или только материнского аллеля (HI9 и CDKN1C).
Ген IGF2 кодирует инсулиноподобный фактор роста, а ген CDKN1C — супрессор
клеточного цикла, ограничивающий деление и рост клеток. Транскрипция генов Н19 и
KCNQOT1 подавляет экспрессию материнской копии IGF2 и отцовской копии CDKN1С
соответственно.
Несбалансированная экспрессия импринтированных генов в регионе 11p15.5 может
происходить по множеству механизмов [6]. 50 % спорадических случаев синдрома
Беквита – Видемана ассоциированы с потерей метилирования гена KCNQOT1 на
материнской хромосоме. В 20 % спорадических случаев причиной заболевания становится
снижение экспрессии материнской копии CDKN1C и повышение экспрессии IGF2,
вызванное отцовской однородительской дисомией. Мутации в материнском аллеле гена
CDKN1C обнаруживают в 5–10 % спорадических случаев и у 40 % семей с аутосомно-
доминантным синдромом Беквита – Видемана. 5 % случаев синдрома связано с
гиперметилированием гена H19 на материнской хромосоме. Менее 1 % случаев
ассоциировано с цитогенетически видимой дупликацией и транслокацией/инверсией
участка 11p15 хромосомы 11 [3].
Также было показано, что при хромосомных перестройках у пациентов с синдромом Беквита-Видемана нарушается ген KCNQ1.
- Ген H19: 5.3-KB DEL
- Ген H19: 1.8-KB DEL
- Ген KCNQ1OT1: DEL
- Ген CDKN1C: 1-BP DEL/2-BP INS, 1086T-AG
- Beckwith J. B. Macroglossia, omphalocele, adrenal cytomegaly, gigantism, and
hyperplastic visceromegaly. Birth Defects Orig. Art. Ser. V(2): 188–196, 1969 - Wiedemann H.-R. Das EMG-Syndrome: Exomphalos, Makroglossie, Gigantismus und
Kohlenhydratstoffwechselstoerung. Z. Kinderheilk. 106: 171–185, 1969 [PubMed: 5797233] - Федеральные клинические рекомендации по диагностике и лечению синдрома
Беквита-Видемана. Москва – 2017 - Kosseff A. L., Herrmann J., Opitz J. M. The Wiedemann-Beckwith syndrome: genetic
consideration and a diagnostic sign. (Letter) Lancet 299: 844 only, 1972. Note: Originally
Volume I [PubMed: 4111600] - Ситдикова И. В., Закиров К. З., Измайлова А. Х. Синдром Видемана-Беквита у
девочки в возрасте 2 месяцев // Казанский медицинский журнал. – 2009. – Т.90. — № 3. – С.
458–459 - Безуглая А. А. Синдром Беквитта-Видемана // Материалы X Международной
студенческой научной конференции «Студенческий научный форум» - Matsuoka S., Edwards M. C., Bai C., Parker S., Zhang P., Baldini A., Harper J. W.,
Elledge S. J. p57(KIP2), a structurally distinct member of the p21(CIP1) Cdk inhibitor family, is
a candidate tumor suppressor gene. Genes Dev. 9: 650–662, 1995.[PubMed: 7729684] - Mitsuya K., Meguro M., Lee M. P., Katoh M., Schulz T. C., Kugoh H., Yoshida M. A.,
Niikawa N., Feinberg A. P., Oshimura M. LIT1, an imprinted antisense RNA in the human
KvLQT1 locus identified by screening for differentially expressed transcripts using
monochromosomal hybrids. Hum. Molec. Genet. 8: 1209–1217, 1999. Note: Erratum: Hum.
Molec. Genet. 8: 1585 only, 1999.[PubMed: 10369866]
Источник