При синдроме гурлера тип наследования

Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
Мукополисахариды или гликозаминогликаны — полисахаридные цепи, синтезируемые клетками соединительной ткани как нормальный компонент многих тканей. Они состоят из большого числа дисахаридных блоков; отличительная черта конкретного гликозаминогликана — природа двух молекул сахаров.
Разложение этих макромолекул происходит в лизосоме и требует пошагового удаления блока моносахаридов в конце цепи с помощью фермента, специфичного для моносахарида и вовлеченной цепи. Таким образом, для разложения любого гликозаминогликана необходима серия ферментов, и один фермент часто участвует в распаде более чем одного гликозаминогликана.
Мукополисахаридозы — разнородная группа, включающая более дюжины болезней накопления, при которых в результате недостатка одного из ферментов, необходимого для их разложения, в лизосомах накапливаются мукополисахариды. При конкретных мукополисахаридозах могут накапливаться один или несколько гликозаминогликанов, если отсутствует фермент, необходимый для их распада.
Недеградировавшие гликозаминогликаны появляются в моче, где они могут быть обнаружены скрининговыми тестами.
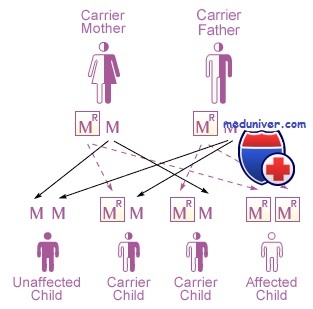
Первые два описанных мукополисахаридоза — Х-сцепленный рецессивный синдром Хантера и более тяжелый аутосомно-рецессивный синдром Гурлер. Каждое из этих заболеваний сначала называли гаргоилизмом из-за грубости черт лица больных детей. Больные дети имеют скелетные аномалии и низкий рост, умственную задержку, а также другие аномалии.
Синдром Гурлер — следствие выраженной недостаточности а1-идуронидазы. Клинически отличное заболевание — синдром Шейе — первоначально считали вызванным мутацией в другом локусе, в основном из-за значительно более мягкого фенотипа. Тем не менее синдромы Шейе и Гурлер оказались аллельными, а мутации а1-идуронидазы, вызывающие синдром Шейе, связаны с более высокой остаточной активностью.
Примеры мукополисахаридозов:
1. Гурлер. Диагностируется рано (<18 мес), помутнение роговицы, скелетные изменения, гепатоспленомегалия, грубое лицо, смерть до 10-летнего возраста. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивное. Из-за аллелей, которые существенно нарушают активность фермента.
2. Шейе. Начало после 5 лет жизн, нормальный интеллект и продолжительность жизни, роговичные помутнения, клапанные пороки сердца. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивный. Очевидно, из-за аллелей, которые оставляют некоторую остаточную активность фермента.
3. Хантера. Гурлер-подобный синдром, но с медленным течением. Дефект фермента: Идуронатсульфатаза. Наследование: Х-сцепленный рецессивный. Мягкий фенотип с переменным поражением цнс.
Разные типы наследования аутосомного синдрома Гурлер и Х-сцепленного синдрома Хантера указывают, что они возникли вследствие мутаций в разных генах. Это различие также показано на клеточных культурах. Хотя фибробласты пациентов при обеих болезнях накапливают мукополисахариды в культуральной среде, накопление корректируется при совместном культивировании обоих типов клеток в одной культуральной посуде.
Коррекция происходит вследствие захвата фибробластами с недостаточностью a-L-идуронидазы при синдроме Гурлер нормальной a-L-идуронидазы, синтезированной фибробластами синдрома Хантера; в клетках с синдромом Хантера происходил обратный феномен. Этот простой эксперимент оказался красивой иллюстрацией того, что две болезни повреждают разные белки. Явление, когда продукт генома одного мутантного гена может скорректировать биохимический дефект у другого, называется генетической комплементацией, а исследования, используемые для обнаружения генетической комплементации, называют анализом комплементации.
Способность клетки усваивать недостающий лизосомный фермент из внеклеточной жидкости — один из механизмов коррекции биохимического дефекта при болезнях накопления после пересадки пациентам нормальных клеток (выделяющих фермент). Радикальный терапевтический эффект получен при лечении некоторых пациентов с мукополисахаридозами, включая синдром Гурлер, при пересадке костного мозга.
Способность клеток усваивать лизосомные ферменты из внеклеточной жидкости также дает обоснование для заместительной ферментной терапии при многих болезнях — стратегия, в значительном числе случаях оказавшаяся в высшей степени эффективной.
— Также рекомендуем «Генетика I-клеточной болезни. Молекулярные основы»
Оглавление темы «Генетика ферментопатий»:
- Генетика фенилкетонурии. Наследование
- Генетика болезни Тея-Сакса. Наследование
- Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
- Генетика I-клеточной болезни. Молекулярные основы
- Генетика менделирующей восприимчивости к грибковой и бактериальной инфекции — mendelian susceptibility to mycobacterial disease (MSMD)
- Генетика гомоцистинурии. Наследование
- Генетика нарушений обмена витаминов (кофакторов). Наследование
- Генетика ферментопатий (ферментативной недостаточности). Наследование
- Генетика недостаточности альфа-1-антитрипсина. Наследование
- Генетика острой перемежающейся порфирии. Наследование
Источник
Одно из редких наследственных генетических заболеваний, которое неуклонно прогрессирует и рано приводит к тяжелой инвалидности – это синдром Гурлера. Характеризуется оно нарушением метаболизма, в результате которого в организме больного постоянно накапливаются продукты обмена. Они разрушают нервную систему, внутренние органы и скелет. Эти разрушения являются необратимыми, поэтому раньше синдром Гурлера был неизлечим, пациенты умирали, едва дожив до 10 лет.
Но при современных возможностях медицины и своевременно начатом лечении возможно устранить патологию обменных процессов и вернуть пациента к нормальной жизни. Правда, уже возникшие деформации скелета и болезни внутренних органов остаются, поэтому такие больные чаще всего все же остаются инвалидами, и у них наблюдаются характерные аномалии внешности.
Характеристика заболевания
Синдром Гурлера – это одна из разновидностей мукополисахаридоза, генетического заболевания, связанного с нарушением выработки определенных лизосомных ферментов. У больных с рождения не производится альфа- L-идуронидаза. Этот фермент ответственен за расщепление определенных групп жиров и углеводов на более простые молекулы. В результате в крови и клетках накапливаются мукополисахариды, которые в здоровом организме разрушаются этим ферментом.
Это вызывает дисфункции разных органов, неправильное развитие тканей. Нерасщепленные мукополисахариды откладываются в мозговых оболочках, внутренних органах, нервных клетках, хрящах, роговице глаза. Страдают из-за этого нервная система, сердце, головной мозг, органы дыхания, глаза, печень, кости и суставы. При рождении дети с синдромом Гурлера кажутся нормальными, симптомы патологии начинают проявляться к концу первого года жизни. Постепенно мукополисахариды накапливаются, нарушения прогрессируют, и без лечения заболевание к 10 годам приводит к смерти пациента.
Причины
Одним из самых серьезных наследственных заболеваний считается мукополисахаридоз, синдром Гурлера от его разновидностей отличается наиболее тяжелым течением. Вызывается болезнь генетической мутацией, которая возникает в хромосоме 4. Тип наследования ее аутосомно-рецессивный. Это означает, что ребенок заболеет только в том случае, если оба родителя являются носителями аномального гена. Причем, у них признаков болезни может не быть. Но с вероятностью в 25% ребенок получит именно дефектный ген. Заболевание это очень редкое, встречается в 1 случае из 100 тысяч новорожденных. С одинаковой частотой патология бывает как у мальчиков, так и у девочек.

Это заболевание наследственное, поэтому первые признаки его проявляются сразу после рождения
Симптомы
Проявления заболевания различаются в зависимости от тяжести его течения. Различают три степени мукополисахаридоза 1 типа. При каждой разновидности симптомы могут быть более или менее выражены, ограничиваться только костными деформациями или отражаться на работе внутренних органов и нервной системы. Причем, все эти патологии с каждым годом жизни пациента все прогрессируют.
Самая тяжелая, отличающаяся быстрым прогрессированием и необратимыми деформациями – это собственно синдром Гурлера. Все признаки начинают проявляться еще в раннем детстве, причем они очень сильно выражены. Характерным отличием от других разновидностей является сильное отставание в умственном развитии и явные аномалии внешности. Смерть наступает обычно до 10 лет от серьезных нарушений работы внутренних органов.
Наиболее легкая форма мукополисахаридоза 1 типа называется синдромом Шейе по имени врача, впервые описавшего патологию. Больные могут вести нормальную жизнь, у них сохраняется интеллект. Но наблюдаются патологии суставов, сердечно-сосудистой системы, нарушения зрения и слуха, характерное изменение внешности.
Промежуточное место занимает синдром Гурлера-Шейе. Он объединяет в себе признаки обоих разновидностей заболевания. Характеризуется патология нормальным интеллектом, но множественными деформациями в костной системе.

У пациентов после 2-3 лет заболевание можно обнаружить по характерным внешним признакам
Внешность пациентов
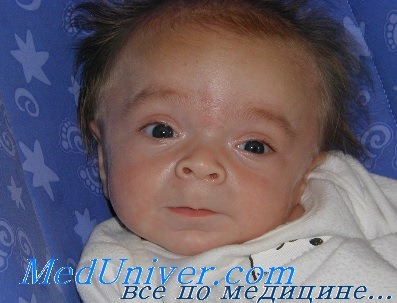
Первым симптомом заболевания является дисморфизм лица. Это огрубление черт по типу гаргоилизма начинается еще с 3-6 месяцев, а со временем только прогрессирует. Поэтому в самых тяжелых формах болезни диагноз можно поставить по фото пациента. Особенно ярко все аномалии внешности видны к 3 годам. Это, прежде всего, увеличенный череп с сильно выраженными швами и лобными буграми, короткая шея, низкий рост, деформации костей разной степени тяжести.
Черты лица ребенка также обладают характерными признаками. Они становятся к году все более грубыми. Переносица вдавленная и широкая, ноздри вывернуты, а кончик носа неестественно увеличен. Глаза широко расставлены и кажутся немного выпуклыми из-за того, что глазницы маленькие. Губы полные, язык такой большой, что не помещается во рту. Поэтому рот у больного постоянно в полуоткрытом положении. Зубы развиваются неправильно, растут нерегулярно, быстро подвергаются кариесу.
Деформации скелета
При этом заболевании почти всегда имеют место деформации скелета ребенка. Прежде всего, это видно на изменении формы грудной клетки. Ребра расширяются, и она приобретает бочкообразную форму. Примерно к году у всех детей с синдромом Гурлера развиваются дисплазии суставов и мягкие аномалии костей. Позвоночник часто искривляется, появляется кифоз, чаще всего в поясничном или грудном отделе. Позвонки выступают и приобретают характер «рыбьих». Таз недоразвит, из-за малого размера головок бедренных костей возникает дисплазия тазобедренных суставов.
Часто у пациентов развивается кистевой туннельный синдром или аномалии развития кисти по типу «когтистой лапы». Пальцы довольно короткие и плохо гнутся. Патологии мелких и крупных суставов проявляются их тугоподвижностью, частыми болями уже в раннем возрасте. Может наблюдаться вальгусная деформация стоп. У больных низкий рост, укороченная шея. Из-за неправильного телосложения они могут ходить только на цыпочках, да еще на полусогнутых ногах. Кроме того, у пациентов наблюдается мышечная гипотония и двигательная заторможенность, которая с возрастом только усиливается.

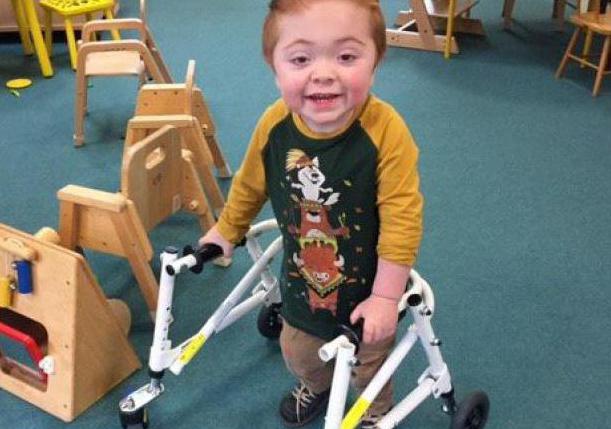
Серьезные деформации скелета, отставание в умственном и физическом развитии часто приводят к инвалидности
Внутренние органы
Дефицит фермента, ответственного за расцепление некоторых углеводов, особенно отражается на состоянии печени и селезенки. У больных уже в раннем детстве эти органы сильно увеличены, поэтому живот кажется неестественно большим. При прогрессировании заболевания может развиться цирроз печени. Страдает также сердечно-сосудистая система. Чаще всего возникает патология клапанов аорты, поэтому наблюдаются шумы в сердце. Может также развиться гипертензия, сужение коронарных сосудов или даже инфаркт.
Из-за сниженного иммунитета такие дети часто подвержены инфекционным заболеваниям. Страдают в основном органы дыхания, часто развивается дыхательная недостаточность, по ночам может наблюдаться апноэ. Характерным признаком болезни может быть также шумное дыхание пациента.
Нервная система
На втором году жизни пациента становятся заметными нарушения интеллектуального развития. Примерно до года ребенок развивается нормально, но потом начинает отставать от своих сверстников, может потерять уже приобретенные навыки. Нарушения проявляются также в речевом развитии. Это связано не только с умственной отсталостью, но и с патологиями речевого аппарата, а также с прогрессирующей глухотой.
Другие патологии
Одним из первых симптомов патологии является появление паховых и пупочных грыж у ребенка. Часто накопление мукополисахаридов в крови вызывает помутнение роговицы или развитие глаукомы. Оно может начаться еще на первом году жизни и быстро прогрессирует, приводя к слепоте. Страдает также орган слуха – у многих пациентов развивается прогрессирующая нейросенсорная тугоухость, часто возникают отиты. У больных с синдромом Гурлера меняется структура ногтей, волосы становятся жесткими, кожа – толстой и грубой.

Часто у детей с синдромом Гурлера развивается помутнение роговицы или даже глаукома
Диагностика
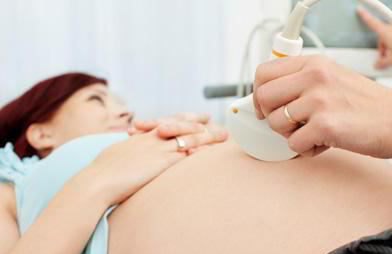
Заболевание это наследственное, поэтому профилактика его невозможна. Но можно провести генетический анализ амниотической жидкости во время внутриутробного развития. Это делается, если в роду у родителей были случаи заболевания. При подозрении на синдром Гурлера после рождения проводятся такие исследования:
- при анализах крови определяется уровень L-идуронидазы;
- в пробах мочи обнаруживается повышенный уровень мукополисахаридов;
- иногда делается генетический анализ крови;
- в более старшем возрасте назначается рентгенография для определения степени деформаций скелета;
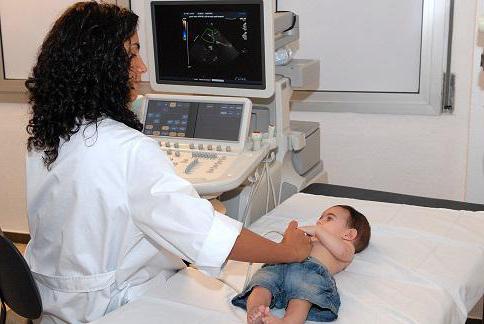
- эхокардиография поможет выявить нарушения работы сердечно-сосудистой системы.
При своевременной диагностике заболевания возможно затормозить серьезные деформации скелета и нарушения работы внутренних органов.
Лечение
При легкой форме патологии симптомы проявляются, как правило, только в подростковом возрасте. Болезнь при этом протекает не так тяжело, поэтому поддается консервативному лечению. Пациента наблюдают многие врачи, которые проводят симптоматическую терапию. У ортопеда лечатся деформации скелета, удаляются грыжи, исправляются патологии суставов. Педиатр купирует проявления вирусных инфекций, следит за состоянием сердечно-сосудистой системы.
Но при тяжелой форме синдрома Гурлера чаще всего основным методом терапии является трансплантация костного мозга или пуповинной крови от здорового совместимого донора. При этом клетки крови больного заменяются на здоровые, которые не содержат генетического дефекта. Поэтому дефицита фермента уже не обнаруживается. Накопление вредных продуктов обмена прекращается, но уже полученные разрушения восстановить не удается. Поэтому очень важно, чтобы операция была проведена как можно раньше. Обычно трансплантацию делают на втором году жизни.
При успешной операции продолжительность жизни больного и ее качество существенно увеличивается. Неврологические нарушения и отставания в интеллектуальном развитии устраняются, но костные деформации и нарушения зрения часто сохраняются.

Если проведение трансплантации невозможно, улучшить состояние больного можно с помощью внутривенного введения специальных препаратов
При заболевании легкой тяжести помочь может также медикаментозная терапия. Применяются препараты, заменяющие нужный фермент. Самым эффективным считается «Альдуразим». Его вводят внутривенно, и такое лечение улучшает состояние всех органов и систем, кроме головного мозга. Это происходит из-за наличия гематоэнцефалитического барьера, который не пропускает этот препарат. «Альдуразим» часто применяется также при тяжелом течении заболевания перед операцией по трансплантации костного мозга, чтобы улучшить состояние пациента.
При консервативном лечении пациента с синдромом Гурлера применяются глюкокортикостероиды, например, «Преднизолон», а также «Кортикотропин», «Тиреоидин», «Декстран», витамин А в больших количествах и сердечные препараты. При невозможности выполнить трансплантацию костного мозга можно делать переливание плазмы крови. Показано также физиотерапевтическое лечение, которое улучшает состояние пациента. Эффективен электрофорез с «Лидазой» на суставы, магнитотерапия, парафин, лазеролечение, массаж, лечебная физкультура.
Это тяжелое наследственное заболевание только с развитием медицины стало возможно вылечить. Но для этого важно как можно раньше поставить диагноз, а родители должны выполнять все предписания врача. Если они проявят терпение и настойчивость, ребенок может развиваться более или менее нормально, общаться со сверстниками и самостоятельно обслуживать себя.
Источник
Мукополисахаридоз – общее название ряда редких заболеваний, которые носят генетический характер. Патология развивается вследствие нехватки в организме некоторых ферментов, помогающих расщеплять жиры и углеводы на простые молекулы. В данной статье рассмотрен мукополисахаридоз 1 типа — синдром Гурлера.
Причины
Болезнь является наследственной по аутосомно-рецессивному типу. Она развивается по причине аномалий обмена мукополисахаридов.
Патогенез
Мукополисахаридоз относится к так называемым лизосомным болезням накопления. В результате дефицита лизосомных ферментов затрудняется катаболизм гликозаминогликанов. Они накапливаются в тканях и органах, нарушая работу организма и его систем. В первую очередь происходит поражение скелета и задержка физического развития.
Внешние признаки и симптомы заболевания
Симптомы заболевания проявляются в виде дефектов костной, соединительной, хрящевой тканей. Основной признак – задержка роста. Этот симптом можно обнаружить раньше остальных, обычно уже к концу первого года жизни становится понятно, что ребенок отстает в росте.
Также мукополисахаридоз можно предположить, видя грубые черты лица. У больных большой язык, гипертелозирм (слишком большое расстояние между парными органами, в данном случае между глазами), деформированные ушные раковины, лоб нависает, зубы искривлены.
К симптомам мукополисахаридоза относятся деформации грудной клетки, выраженный кифоз грудопоясничного отдела позвоночника. При проведении рентгена можно обнаружить преждевременное окостенение затылочно-теменного шва без нарушения ядер окостенения.
В большинстве случаев болезнь сопровождает ограничение подвижности суставов, абдоминальные грыжи, гепатоспленомегалия (увеличение печени и селезенки из-за патологических процессов, происходящих в результате заболевания).
Со стороны неврологии отмечаются двигательная заторможенности и мышечная гипотония. Также при мукополисахаридозе происходит ослабление слуха и снижение интеллекта вплоть до тяжелого слабоумия. Вследствие прогрессирующих системных поражений скелета внутренние органы также подвергаются нарушениям в различной степени.
Типы мукополисахаридоза
Различают несколько типов заболевания, которые различаются выраженностью костных изменений и нарушений психики:
- I — синдром Гурлера.
- II — синдром Гунтера (Хантера).
- III — синдром Санфилиппо.
- IV — синдром Моркио.
- VI — синдром Марото – Лами.
- VII — синдром Слая.
Деление в медицинских практиках разных стран может отличаться. В тип V обычно выделяют синдром Шейе. В американском же сообществе больных мукополисаридозами делят по тяжести проявления симптоматики первого типа и выделяют три фенотипа: синдром Гурлера, синдром Шейе и промежуточный между ними синдром Гурлер – Шейе (из них Гурлер самый тяжелый, Шейе – самый легкий).
Синдром Гурлера
Эта форма встречается чаще остальных и была описана ранее других синдромов. К тому же клиническая картина наиболее яркая и типичная из всех видов мукополисахаридоза.
Синдром Гурлера развивается в результате аутосомно-рецессивного наследования. Этот тип болезни характеризуется очень быстрым прогрессом. Несмотря на то что мукополисахаридоз первого типа схож со вторым (Гунтера, или Хантера), это более сложное заболевание. Впервые данная форма была описана в 1919 году Гертрудой Гурлер (поэтому правильное название – синдром Гурлер, а не Гурлера). Частота появления – один случай на 20-25 тысяч человек, причем в большинстве случаев родители заболевшего ребенка находятся в кровном родстве. Поэтому если ставится диагноз «синдром Гурлера», причины необходимо искать на генетическом уровне. Симптомы проявляются практически сразу после рождения, а к году-двум клиническая картина уже выражена полностью.
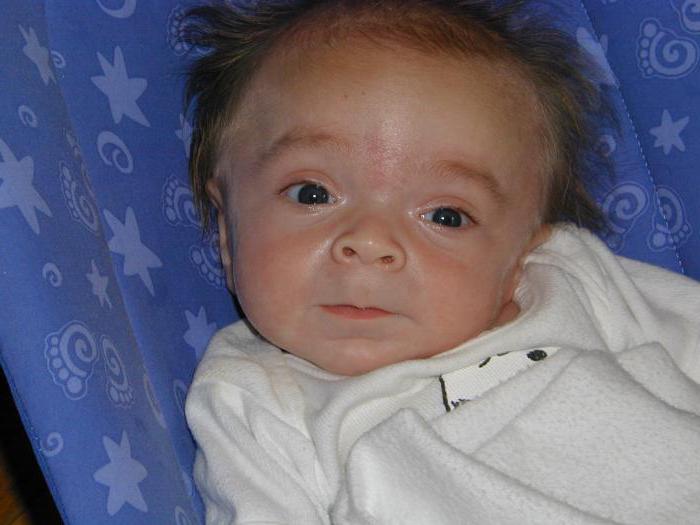
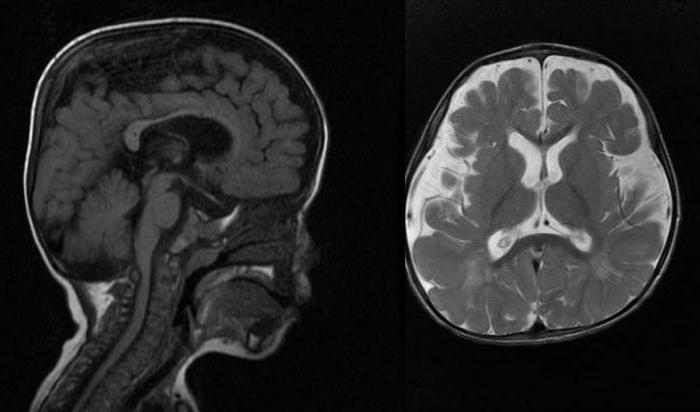
Синдром Гурлера – классическое проявление заболевания. По мере развития недуга замедляется рост, отмечается видимое помутнение роговицы, сосуды носа переполняются кровью. При этой форме заболевания при рентгенологическом исследовании можно обнаружить расширение турецкого седла, укорочение и расширение длинных костей, гипоплазию и заостренность позвонков поясничной области (так называемые рыбьи позвонки), деформации позвоночного столба (больные страдают кифозом и лордозом грудопоясничной области позвоночника). Начинаются патологии сердечно-сосудистой системы — закупориваются коронарные артерии, изменяются клапаны, миокард, эндокард, сердце увеличивается в размерах.
Отмечается гидроцефалия, причиной которой становятся отложения мукополисахаридов в мозговых оболочках. Определяются очаги демиелинизации. Мукополисахариды также откладываются в печени, селезенке, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, хрящах.
Дети рождаются уже с характерным внешним видом — у них очень своеобразные, грубые черты лица, из-за чего другое название мукополисахароидозов – гаргоилизм (от слова «гаргулья» — фантастическая фигура с необычными чертами лица), в том числе так называют и синдром Гурлера. Фото, на которых изображены больные, наглядно иллюстрируют причудливое искажение черт лица ребенка. У таких детей изменен череп – он приобретает форму киля лодки, наблюдается так называемая скафоцефалия, запавшая переносица, толстые губы, большой язык, крутой лоб, короткая шея и характерное выражение лица. Внешне это действительно напоминает то, как изображают мифологических гаргулий.

Также у них укорочена грудная клетка, нижние ребра выступают, есть признаки кифоза, суставы (особенно пальцев и локтей) малоподвижны, могут быть паховые и пупочные грыжи. Ногти могут приобретать вид часовых стекол, волосы становятся жесткими и сухими, голос – низким и хриплым. Вероятна тугоухость или даже глухота. Больные часто страдают от кариеса, который провоцирует синдром Гурлера.
Симптомы включают патологии дыхательной системы, из-за этого ребенок дышит ртом, у него появляются аденоиды, он подвержен вирусным инфекциям. Со временем у него развиваются характерные для мукополисахаридозов проблемы с печенью и селезенкой (в результате увеличен живот), слабоумие.
Рост остается карликовым. Из-за неправильного телосложения и деформации позвоночника больные ходят на полусогнутых ногах, на цыпочках.
У синдрома Гурлера злокачественный прогрессирующий характер, поэтому крайне быстро наступает инвалидизация больных. Многие не доживают даже до 10 лет.
Диагностика
Пациенту необходимо провести клиническое, рентгенологическое, биохимическое, генеалогическое, а также молекулярно-генетические исследования. Диагностика проводится по клиническим проявлениям болезни, на основе рентгенологических исследований и анализа мочи, по которому определяется активность ферментов и экскреция гликозаминогликанов.
Лечение мукополисахаридоза
Если больному поставлен диагноз «синдром Гурлера», лечение предполагается больше симптоматическое. Пациент наблюдается комплексно у ортопеда, хирурга, педиатра, отоларинголога, нейрохирурга, офтальмолога и невропатолога. Больному проводится ортопедическая коррекция нарушений опорно-двигательного аппарата, удаляют грыжи, лечатся часто возникающие у таких пациентов вирусные заболевания, нарушения слуха, отиты, синуситы. Также под наблюдение попадает сердечно-сосудистая система.
Используются гормональные препараты, которые временно улучшают состояние пациента:
- глюкокортикоиды,
- кортикотропин,
- тиреоидин.
Помимо этого больному показаны витамин А, декстран 70, которые также временно улучшают состояние больного. Недолгосрочное улучшение дает переливание препаратов плазмы крови.
При синдроме Гурлера пациенту может быть назначено физиотерапевтическое лечение: электрофорез лидазы на область пораженных суставов, лазерная пунктура, магнитотерапия, парафиновые аппликации. Также больным рекомендуется заниматься лечебной физкультурой, упражнения которой воздействуют на суставы и позвоночник. Хорошие результаты часто дает массаж.
Так как больные синдромом Гурлера подвержены респираторным заболеваниям, необходимо своевременно проводить санацию очагов инфекций во рту и носоглотке.
Для лечения мукополисахаридоза 1-го типа часто проводятся хирургические вмешательства – пересадка роговицы и коррекция клапанных пороков сердца и ущемлений нервов. В международной практике помимо симптоматического лечения медикаментами, физиотерапией или хирургических вмешательств используется заместительная ферментная терапия, а также трансплантация стволовых клеток.
При необходимости больному проводят грыжеиссечения, удаление аденоидов, антиглаукоматозные операции, трахеостомию, протезирование тазобедренных суставов, шунтирование при гидроцефалии и пр.
Прогноз
Прогноз неблагоприятен как для синдрома Гурлера, так и для других форм, которые имеет мукополисахаридоз. Синдром Гурлера отличается наибольшей безнадежностью. Изменения скелета с каждым годом увеличиваются, в результате органы и системы подвергаются более значительным нарушениям. Если ребенок не умирает от пневмонии в раннем возрасте, то к 7-12 годам это уже беспомощный физически и психически инвалид. До юношеского возраста доживают единицы.
Профилактика
Предупредить данное заболевание невозможно. Но можно обнаружить его на самой ранней стадии – при пренатальной диагностике. Для этого проводится анализ амниотических клеток на предмет дефицита фермента (в случае положительного результата беременной рекомендуется аборт).
За счет ранней диагностики и своевременной терапии развившейся компрессии спинного мозга можно избежать необратимых повреждений нервов. Для профилактики обязательно проводится медико-генетическая консультация.
Прогнозы лечения
Несмотря на сложности в плане лечения синдрома Гурлера, в последние 20 лет во многих развитых странах используется трансплантация костного мозга, которая значительно улучшает качество жизни больных. Более 10 лет применяются препараты заместительной терапии для лечения всех проявлений мукополисахаридоза не неврологического характера.
Источник