Полигландулярный аутоиммунный синдром код по мкб

Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Аутоиммунный полигландулярный синдром.
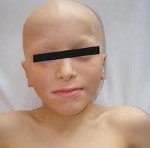
Аутоиммунный полигландулярный синдром
Описание
Аутоиммунный полигландулярный синдром. Эндокринопатия аутоиммунного генеза, протекающая с одновременным первичным множественным поражением желез внутренней секреции и других органов. При аутоиммунном полигландулярном синдроме 1 типа отмечается надпочечниковая недостаточность, кандидоз кожи и слизистых, гипопаратиреоз; аутоиммунный полигландулярный синдром 2 типа протекает с развитием надпочечниковой недостаточности, гипер- или гипотиреоза, инсулинозависимого сахарного диабета, первичного гипогонадизма, миастении, стеатореи и тд нарушений. Диагностика включает определение комплекса лабораторных показателей (биохимического анализа, гормонов крови и мочи), УЗИ и КТ надпочечников, УЗИ щитовидной железы. Лечение аутоиммунного полигландулярного синдрома требует назначения заместительной терапии гормонами (кортикостероидами и минералокортикоидами, L-тироксином).
Дополнительные факты
Аутоиммунный полигландулярный синдром (АПГС) – иммуноэндокринное нарушение, характеризующееся первичной функциональной недостаточностью нескольких желез внутренней секреции, а также неэндокринными органоспецифическими заболеваниями. В эндокринологии различают аутоиммунный полигландулярный синдром 1 и 2 типов (АПГС-1, 2), которые имеют свои генетические, иммунологические и клинические особенности.
Аутоиммунный полигландулярный синдром 1 типа обычно манифестирует в детском возрасте (10-12 лет) и иногда обозначается в литературе термином «ювенильная семейная полиэндокринопатия». В целом распространенность АПГС-1 невелика, однако заболевание несколько чаще встречается в мужской популяции, преимущественно у жителей Финляндии, Сардинии, Ирана, что объясняется длительной генетической обособленностью этих народов. Аутоиммунный полигландулярный синдром 1 типа включает триаду признаков: надпочечниковую недостаточность, гипопаратироз и кандидамикоз.
Наиболее частым вариантом множественной эндокринопатии аутоиммунной природы является аутоиммунный полигландулярный синдром 2 типа, который развивается у взрослых (старше 20-30 лет); среди заболевших преобладают женщины. Компонентами АПГС-2 являются надпочечниковая недостаточность, сахарный диабет 1-го типа, аутоиммунное поражение щитовидной железы по типу первичного гипотиреоза или тиреотоксикоза. Кроме эндокринных нарушений, аутоиммунному полигландулярному синдрому 1 и 2 типов сопутствуют другие органоспецифические проявления.
Аутоиммунный полигландулярный синдром
Симптомы
Гиперкальциемия. Лейкоцитоз. Судороги.
Причины
Оба типа аутоиммунного полигландулярного синдрома являются генетически детерминированными, на что указывает семейно-наследственный характер заболевания. При этом АПГС-1 поражаются братья и сестры одного поколения; аутоиммунным полигландулярным синдромом 2 типа – представители одной семьи на протяжении нескольких поколений.
Аутоиммунный полигландулярный синдром 1 типа – единственное известное аутоиммунное заболевание, имеющее моногенную природу. К развитию АПГС-1 приводит мутация гена аутоиммунного регулятора (AIRE), расположенного на длинном плече 21 хромосомы (21q22. 3). Аутоиммунный полигландулярный синдром-1 наследуется по аутосомно-рецессивному типу и не связан с гаплотипами HLA.
Аутоиммунный полигландулярный синдром 2 типа ассоциирован с HLA-гаплотипом (антигенами DR3, DR4, DR5, В8, Dw3). Вероятно, что механизм развития АПГС-2 связан с аномальной экспрессией антигенов HLA-системы на клеточных мембранах желез внутренней секреции, которая запускается под воздействием каких-либо внешних факторов.
Диагностика
Критериями для постановки клинического диагноза служат подтвержденные лабораторным и инструментальным путем изолированные компоненты аутоиммунного полигландулярного синдрома (кожно-слизистый кандидоз, гипопаратиреоз и ХНН при АПГС-1; ХНН, аутоиммунный тиреоидит и сахарный диабет при АПГС-2). При АПГС 1 типа наиболее показательно проведение молекулярно-генетического анализа, позволяющего выявить характерную генную мутацию.
Лабораторное обследование при аутоиммунном полигландулярном синдроме включает определение биохимических показателей крови (уровня общего и ионизированного кальция, фосфора, калия, натрия, билирубина, трансаминаз, щелочной фосфатазы, общего белка, мочевины, креатинина, глюкозы), КОС крови и тд Важные диагностические сведения дают результаты исследований гормонов: ТТГ, свободного тироксина, паратгормона, АКТГ, инсулина, С-пептида, кортизола, ренина, альдостерона, соматомедина С, АТ-ТПО, АТ к бета-клеткам поджелудочной железы и GAD, тестостерона, ЛГ, ФСГ и тд.
Инструментальные исследования включают УЗИ брюшной полости, щитовидной железы, органов малого таза (у женщин) и мошонки у мужчин, ЭхоКГ, КТ надпочечников. При наличии показаний проводятся консультации узких специалистов – диабетолога, гастроэнтеролога, гепатолога, дерматолога, миколога, невролога, гематолога, офтальмолога, гинеколога-эндокринолога, андролога, ревматолога.
Лечение
Терапия аутоиммунного полигландулярного синдрома представляет сложную задачу и складывается из лечения его отдельных компонентов. Основу патогенетической терапии составляет постоянная заместительная гормонотерапия при функциональной недостаточной пораженных эндокринных желез. При надпочечниковой недостаточности назначаются глюкокортикоиды (гидрокортизон, дексаметазон, преднизолон, триамцинолон), минералокортикоиды (ДОКСА, триметилацетат дезоксикортикостерона и тд ), при гипотиреозе — L-тироксин.
При кандидамикозе, сопровождающем аутоиммунный полигландулярный синдром 1 типа, используются противогрибковые препараты. При сахарном диабете на фоне аутоиммунного полигландулярного синдрома 2 типа может потребоваться иммуносупрессивная терапия циклоспорином. Пациентам рекомендуется употребление повышенного количества аскорбиновой кислоты и соли. Запрещается прием алкоголя, некоторых лекарственных препаратов.
Прогноз
Раннее выявление аутоиммунного полигландулярного синдрома и проведение адекватной заместительной терапии позволяют контролировать течение заболевания. Однако трудоспособность, как правило, снижается – пациентам присваивается II-III группа инвалидности.
Пациенты с аутоиммунным полигландулярным синдромом подлежат диспансерному наблюдению эндокринолога и других специалистов. При стрессовых состояниях, интеркуррентных инфекциях, физическом или умственном перенапряжении больным необходимо увеличение дозы гормонов. Необходимо немедленное обращение к врачу при любом ухудшении самочувствия. Летальный исход при аутоиммунном полигландулярном синдроме может наступать от ларингоспазма, висцерального кандидамикоза, острой надпочечниковой недостаточности.
Основные медуслуги по стандартам лечения | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: E31.0
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E20-E35 Нарушения других эндокринных желез / E31 Полигландулярная дисфункция
Определение и общие сведения[править]
Аутоиммунная полигландулярная недостаточность — это заболевания, характеризующиеся аутоиммунным поражением нескольких эндокринных желез и других органов.
Классификация (в порядке убывания распространенности)
Аутоиммунный полигландулярный синдром типа II
Синонимы: синдром Шмидта
В 1926 г. M. B. Schmidt описал двух больных с хроническим тиреоидитом и первичной надпочечниковой недостаточностью (болезнью Аддисона). В дальнейшем были обнаружены другие компоненты синдрома. В настоящее время принято различать следующие компоненты аутоиммунного полигландулярного синдрома типа II:
1. Обязательные
а. Первичная надпочечниковая недостаточность (у всех больных).
б. Поражения щитовидной железы (у всех больных)
1) Хронический лимфоцитарный тиреоидит (у 95—97% больных).
2) Диффузный токсический зоб (у 3—5% больных).
2. Частые
а. Инсулинозависимый сахарный диабет (у 40—50% больных).
б. Первичный гипогонадизм (у 20—30% больных).
в. Миастения (у 20—30% больных).
г. Витилиго (у 15—25% больных).
3. Другие
а. Целиакия.
б. Аутоиммунный гастрит.
в. Алопеция.
г. Синдром мышечной скованности (см. гл. 49, п. II.Ж).
д. Серозиты.
е. Тимома.
Обычно поражает людей в возрасте 20—40 лет. Генетическим маркером предрасположенности к аутоиммунному полигландулярному синдрому типа II является гаплотип HLA-DR3, HLA-DQB1*0201.
Риск развития отдельных компонентов аутоиммунного полигландулярного синдрома типа II (в первую очередь — аутоиммунных поражений щитовидной железы) повышен при других синдромах, связанных с HLA-B8 и HLA-DR3, таких, как синдром Шегрена, изолированный дефицит IgA, дерматомиозит, хронический активный гепатит, герпетиформный дерматит.
Аутоиммунный полигландулярный синдром типа I
Синонимы: MEDAC синдром, синдром APECED
Аутоиммунный полигландулярный синдром 1 типа — редкое заболевание, которое чаще встречается в популяциях с высокими показателями кровного родства, а также в Финляндии в силу эффекта основателя, где показатель распространенности оценивается в 1 /25 000 человек. На северо-западе Франции распространенность оценивается в 1 /500 000 человек.
1. Основные компоненты синдрома: хронический генерализованный гранулематозный кандидоз (кандидоз кожи и слизистых), гипопаратиреоз, первичная надпочечниковая недостаточность. Диагноз устанавливают при наличии любых двух заболеваний.
2. Другие компоненты: первичный гипотиреоз, хронический активный гепатит, синдром нарушенного всасывания, первичный гипогонадизм (первичная надпочечниковая недостаточность), витилиго, аутоиммунный гастрит, алопеция.
3. В отличие от типа II, аутоиммунный полигландулярный синдром типа I обычно возникает уже в грудном возрасте. Первым проявлением его чаще всего бывает кандидоз.
4. Аллели HLA не детерминируют предрасположенность к аутоиммунному полигландулярному синдрому типа I. У некоторых больных имеются мутации гена AIRE на 21-й хромосоме (см. гл. 49, п. I.Б.3).
Этиология и патогенез[править]
1. Все аутоиммунные полигландулярные синдромы генетически детерминированы. Об этом в первую очередь свидетельствует семейный характер этих заболеваний. Например, аутоиммунный полигландулярный синдром типа I поражает большинство братьев и сестер в одном поколении, а типа II — несколько поколений одной семьи.
2. Выявлены гены, определяющие предрасположенность к отдельным компонентам аутоиммунного полигландулярного синдрома. К ним относятся прежде всего гены HLA. Например, у больных аутоиммунным полигландулярным синдромом типа II часто выявляются аллели HLA-DR3, -DR5 и -B8, характерные для аутоиммунных поражений щитовидной железы, а также аллели HLA-DR4 и HLA-DQB1*0302, характерные для поражения бета-клеток.
3. Предполагают, что существуют гены, мутации которых лежат в основе развития аутоиммунного полигландулярного синдрома в целом. Такие гены могут локализоваться как в области HLA, так и в других областях генома. Показано, что при аутоиммунном полигландулярном синдроме типа II часто встречаются аллели HLA-DR3 и HLA-DQB1*0201, причем частоты этих аллелей не зависят от того, какие заболевания входят в состав синдрома. Недавно на 21-й хромосоме был найден ген AIRE, регулирующий аутоиммунитет (21q22.3). Мутации этого гена обнаружены у многих больных аутоиммунным полигландулярным синдромом типа I.
4. Пока не выяснено, почему разные аутоиммунные полигландулярные синдромы включают разные составляющие.
Патогенез. Генетические дефекты обусловливают не сами аутоиммунные полигландулярные синдромы или их компоненты, а предрасположенность к ним. Для проявления этих дефектов требуется воздействие негенетических пусковых факторов. В большинстве случаев многообразные нарушения проявляются не одновременно, а последовательно; иногда с интервалом в несколько лет. Например, первым симптомом аутоиммунного полигландулярного синдрома типа I обычно бывает генерализованный гранулематозный кандидоз (кандидоз кожи и слизистых), а надпочечниковая недостаточность проявляется позже.
Клинические проявления[править]
Аутоиммунная полигландулярная недостаточность: Диагностика[править]
При подозрении на аутоиммунный полигландулярный синдром типа II руководствуются следующими правилами:
1. Соотношение частот отдельных компонентов аутоиммунного полигландулярного синдрома типа II в семьях с этим синдромом такое же, как среди населения, но риск каждого компонента в 10—100 раз выше.
2. Если у родственника больного аутоиммунным полигландулярным синдромом типа II выявлено изолированное аутоиммунное эндокринное заболевание (например, первичная надпочечниковая недостаточность), следует искать другие, скрытые компоненты синдрома. Если в местности, где проживает больной, повышена распространенность инсулинозависимого сахарного диабета, надо исключить его и у больного.
3. Скрытые эндокринные заболевания имеются у 1 из 6 родственников больного аутоиммунным полигландулярным синдромом типа II. Наиболее вероятен гипотиреоз. Поэтому у больных и их родственников нужно оценивать функцию щитовидной железы каждые 5 лет (а при наличии жалоб и симптомов даже чаще).
4. Чтобы выявить больных и родственников с наибольшим риском заболевания, можно типировать аллели HLA и определять органоспецифические аутоантитела, но самым дешевым способом обследования пока остается внимательное изучение анамнеза и физикальное исследование, направленное на выявление признаков заболеваний, входящих в состав синдрома.
Дифференциальный диагноз[править]
Аутоиммунная полигландулярная недостаточность: Лечение[править]
Чтобы предупредить гипоадреналовый криз у больных с нелеченной первичной надпочечниковой недостаточностью и гипотиреозом, стероиды назначают до начала заместительной терапии левотироксином . Считается, что левотироксин ускоряет метаболизм остаточного кортизола и тем самым усугубляет надпочечниковую недостаточность. Главная задача ведения больных аутоиммунным полигландулярным синдромом типа I и II и их родственников — выявление доклинических стадий гипотиреоза, надпочечниковой недостаточности и аутоиммунного гастрита.
Профилактика[править]
Прогнозирование и профилактика
1. Если в семье имеются случаи аутоиммунного полигландулярного синдрома, можно рассчитать риск у родственников больных.
2. Обнаружив у больного одно из заболеваний — компонентов аутоиммунного полигландулярного синдрома, следует заподозрить наличие других скрытых заболеваний.
3. Выявляя генетические и иммунологические маркеры предрасположенности к аутоиммунным заболеваниям — компонентам аутоиммунного полигландулярного синдрома, можно обнаруживать эти заболевания на доклинической стадии и проводить их профилактику.
Прочее[править]
IPEX-синдром
Синонимы: Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии и энтеропатии; аутоиммунная энтеропатия 1-го типа
Определение и общие сведения ( в т.ч. эпидемиология)
Тяжелое врожденное системное аутоиммунное заболевание, характеризующееся рефрактерной диарей, эндокринными нарушениями, поражением кожи и инфекциями.
Распространенность неизвестна. В настоящее время зарегистрировано менее 150 случаев, однако, вероятно, их число недооценено.
Этиология и патогенез
Синдром IPEX вызывается мутациями в гене FOXP3 (Xp11.23). Этот ген кодирует forkhead фактор транскрипции, контролирующий развитие и функцию регуляторных Т-клеток CD4+ CD25+, — основной популяции лимфоцитов, участвующей в обратной регуляции иммунных реакций и аутотолерантности.
Наследуется по рецессивному типу сцепленному с Х-хромосомой. Имеется 50% риск передачи, если плод мужского пола, 50% девочек будут носителями.
Клинические проявления
Обычно развивается в течение первых нескольких дней или недель жизни и поражает исключительно мальчиков. Он проявляется последовательно в виде триады симптомов, состоящей из энтеропатии, аутоиммунных расстройств и поражения кожи, однако клинические признаки и тяжесть заболевания могут значительно отличаться среди пациентов.
Тяжелая аутоиммунная энтеропатия проявляется в виде профузной секреторной диареи с мальабсорбцией, нарушением баланса электролитов, снижением прибавки массы тела и отставанием в росте. Также могут наблюдаться рвота, кишечная непроходимость, гастрит или колит. Кроме того, у пациентов развивается аутоиммунная эндокринопатия, как правило, это инсулинозависимый сахарный диабет (тип 1) и тиреоидит, проявляющийся к гипо- или гипертиреозом. Поражение кожи включает в себя множественные зудящие высыпания, напоминающие экзему, псориаз, и/или атопический или эксфолиативный дерматит. Реже может наблюдаться алопеция или ониходистрофия. У больных также может развиваться аутоиммунная цитопения, тромбоцитопения, гемолитическая анемия и нейтропения. Аутоиммунные нарушения могут приводить к пневмонии, гепатиту, нефриту, миозиту, спленомегалии и/или лимфаденопатии. Возможно присоединений локальных или системных инфекций (таких как пневмония, вызванная инфекцией Staphylococcus aureus, кандидоз), однако, вероятнее всего, они связаны с поражением кожного и желудочно-кишечного барьеров, иммуносуппрессивной терапией и недостаточным питанием, а не первичным иммунодефицитом.
Диагностика
Диагноз ставится на основании клинического обследования, анализа семейного анамнеза и лабораторных исследований, выявляющих аутоиммунную энтеропатию (аутоантитела против энтероцитов, гармонина и виллина), сахарный диабет 1-го типа (антитела против инсулина, островковых клеток поджелудочной железы или глутаматдекарбоксилазы), тиреоидит (антитела против тироглобулина и микросомальные антитела к тиреоидной пероксидазе) и цитопению (антитела против тромбоцитов и нейтрофилов, положительный тест Кумбса).
Диагноз подтверждается с помощью молекулярно-генетического исследования.
Дифференциальный диагноз
Включает в себя: синдром Вискотта-Олдрича и синдром Оменна, дефицит STAT1, дефицит CD25, дефицит IL10R, дефицит STAT5b, преходящий диабет новорожденных, тяжелый комбинированный иммунодефицит или промежуточные формы комбинированного иммунодефицита, Х-сцепленную тромбоцитопению и гипоплазию или врожденное недоразвитие поджелудочной железы.
Пренатальная диагностика образца ворсин хориона может проводиться с помощью анализа кариотипа после определения пола плода в семьях, где выявлена патологическая мутация.
Лечение
Единственным средством лечения IPEX является трансплантация гемопоэтических стволовых клеток (ТГСК), которая наиболее успешна на ранних стадиях заболевания. Поддерживающая терапия включает в себя моно- или комбинированную иммуносупрессивную терапию глюкокортикоидами (преднизолон и метилпреднизолон), циклоспорин А, такролимус, азатиоприн, рапамицин, пожизненный прием инсулина и гормонов щитовидной железы в случае их недостаточности, наружное нанесение циклоспорина А при поражении кожи и парентеральное питание при тяжелых случаях энтеропатии.
Прогноз
При отсутствии своевременной диагностики и лечения, как правило, ведет к летальному исходу в течение первых 2 лет жизни. Некоторые пациенты доживают до детского возраста. С ТГСК вероятная продолжительность жизни будет нормальной, однако трудности с кормлением могут сохраняться в течение многих месяцев после ТГСК. Нейропсихическое развитие нормальное.
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
1. Ahonen P, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 322:1830, 1990.
2. Annesley TM, et al. Artifactual hypercalcemia in multiple myeloma. Mayo Clin Proc 57:572, 1982.
3. Bardwick PA, et al. Plasma cell dyscrasia with polyneuropathy, organomegaly endocrinopathy, M protein and skin changes: The POEMS syndrome. Medicine 59:311, 1980.
4. Combs RM. Malignant thymoma, hypothyroidism and immune disorder. South Med J 61:337, 1968.
5. Eisenbarth GS, Jackson R. The immunoendocrinopathy syndromes. In D Foster, P Wilson (eds), Williams Textbook of Endocrinology (8th ed). Philadelphia: Saunders, 1991.
6. Genovese S, et al. Distinct cytoplasmic islet cell antibodies with different risks for type I (insulin-dependent) diabetes mellitus. Diabetologia 35:385, 1992.
7. Gianani R, et al. Prognostically significant heterogeneity of cytoplasmic islet cell antibodies in relatives of patients with type I diabetes. Diabetes 41:347, 1992.
8. Goldman J, et al. Characterization of circulating insulin and proinsulin-binding antibodies in autoimmune hypoglycemia. J Clin Invest 63:1050, 1979.
9. Kahn CR, Harrison LC. Insulin receptor autoantibodies. In PJ Randle, DF Steiner, WJ Whelan (eds), Carbohydrate Metabolism and Its Disorders (vol III). London: Academic, 1981. Pp. 279.
10. Keller RJ, et al. Insulin prophylaxis in individuals at high risk of type I diabetes. Lancet 341:927, 1993.
11. Moroz LA, et al. Thyroid disease with monoclonal (immunoglobulin G) antibody to triiodothyronine and thyroxine. J Clin Endocrinol Metab 56:1009, 1983.
12. Nagamine K, et al. Positional cloning of the APECED gene. Nat Genet, 174:393, 1997.
13. Neufeld N, et al. Two types of autoimmune Addison’s disease associated with difference polyglandular autoimmune (PGA) syndromes. Medicine 60:355, 1981.
14. Pugliese A, et al. Genetics of susceptibility and resistance to insulin-dependent diabetes in stiff-man syndrome. Lancet 344:1027, 1994.
Действующие вещества[править]
Источник