Пахидермопериостоз код по мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Пахидермопериостоз.
Пахидермопериостоз
Описание
Пахидермопериостоз. Генетически гетерогенное заболевание, которое проявляется комплексным поражением кожных покровов, суставов, костей и ряда внутренних органов. Симптомами этого состояния являются гиперплазия всех слоев кожи, гипергидроз, нарушения со стороны сердечно-сосудистой системы, деформация дистальных фаланг пальцев по типу «барабанных палочек». Диагностика пахидермопериостоза осуществляется на основании данных осмотра пациента, рентгенологических и биохимических исследований, изучения наследственного анамнеза и молекулярно-генетического анализа. Специфического лечения заболевания не существует, но симптоматические мероприятия способны длительное время сдерживать его развитие.
Дополнительные факты
Пахидермопериостоз (первичная гипертрофическая остеоартропатия) – наследственное заболевание с различным (согласно последним открытиям в области генетики) механизмом наследования, поражающее кожные покровы, опорно-двигательный аппарат и сердечно-сосудистую систему. По некоторым данным, этой состояние впервые было описано еще в 1868-м году Г. Фридрайхом, однако его полноценное исследование и определение в качестве отдельной нозологической единицы было осуществлено А. Туреном только в 1935-м году. Основной причиной столь долгой невозможности определить пахидермопериостоз, как самостоятельное заболевание, стало то, что многие его проявления схожи с вторичными (симптоматическими) нарушениями, обусловленными поражением легких и сердца или онкологическими процессами. Данная патология намного чаще поражает мужчин (половое распределение заболевших составляет 8:1) и при этом протекает у них в целом тяжелее, нежели у женщин. Встречаемость пахидермопериостоза в настоящий момент точно не определена – основными препятствиями для этого является частая ошибочная диагностика с симптоматическими формами и наличие стертых вариантов заболевания, наблюдаемое у гетерозигот.
Пахидермопериостоз
Причины
Изучение этиологии и патогенеза пахидермопериостоза продолжалось очень долго, только в 2008 году удалось идентифицировать первый ген, ответственный за развитие этого заболевания. Им является HPGD, расположенный на 4-й хромосоме и кодирующий фермент NAD+-зависимой 15-гидрокси-простогландин дегидрогеназы. Функцией этого белка является дегидратация простагландина Е, процесс считается важным звеном метаболизма и утилизации этого биологически активного вещества. В результате мутации HPGD данный процесс нарушается, концентрация простагландина увеличивается, что и является причиной развития пахидермопериостоза. В настоящий момент среди врачей-генетиков ведется дискуссия по поводу механизма наследования мутаций этого гена – большинство склоняется к тому, что он является аутосомно-рецессивным. Но наличие стертых симптомов пахидермопериостоза у гетерозигот по дефектному гену HPGD дает основания некоторым специалистам заявлять о аутосомно-доминантном наследовании с неполной пенетрантностью. Вариант этой патологии, обусловленный мутацией HPGD, был назван первичным гипертрофической остеоартропатией 1-го типа.
Другая форма пахидермопериостоза была открыта вскоре после идентификации гена HPGD – оказалось, что у некоторых больных с доказанным семейным характером патологии отсутствуют такие генетические дефекты. В итоге была выявлена первичная гипертрофическая остеоартропатия 2-го типа, обусловленная мутацией гена SLCO2A1, локализованного на 3-й хромосоме. Продуктом экспрессии данного гена является трансмембранный белок, облегчающий транспорт молекул простагландинов через биологические мембраны. Нарушение структуры этого протеина, вызванное генетическими дефектами, приводит к ухудшению транспорта простагландинов и их накоплению в кровотоке, что, как и в предыдущем случае, является причиной пахидермопериостоза. Формы этого заболевания, обусловленные мутацией гена SLCO2A1, наследуются по аутосомно-рецессивному механизму.
Классификация
В настоящее время выделяют также малоизученную аутосомно-доминантную разновидность пахидермопериостоза. Она является наиболее редкой, и ее этиология до сих пор неясна – по некоторым данным, это могут быть как варианты мутации HPGD, так и повреждения других, еще не идентифицированных генов. Клинические исследования таких больных (в частности, определение уровня простагландина Е) указывают на то, что патогенез этой формы пахидермопериостоза мало отличается от других типов заболевания, но имеет ярко выраженный аутосомно-доминантный характер наследования.
Симптомы
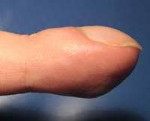
Пахидермопериостоз характеризуется довольно разнообразной клинической картиной, особенно в отношении первых симптомов заболевания. В большинстве случаев проявления патологии можно обнаружить уже в первые несколько лет жизни ребенка, но наиболее выраженными они становятся к подростковому возрасту. Описаны случаи развития симптомов пахидермопериостоза у взрослых людей, которые до этого были фенотипически здоровыми. В случае раннего начала заболевания одним из первых проявлений будет нарушение и задержка зарастания родничков на черепе, в дальнейшем присоединяются дерматологические симптомы. К ним относят увеличение толщины кожных покровов на лице, голове, кистях и стопах, развитие их складчатости, иногда в сочетании с гиперкератозом. Достаточно сильно при пахидермопериостозе страдают веки – происходит утолщение хрящевой основы и кожи, дистрофия конъюнктивы, развитие кист из тарзальных желез. Характерен также выраженный ладонный и стопный гипергидроз.
Замкнутость. Разбитость.
Диагностика
Для определения наличия пахидермопериостоза используют данные осмотра больного, изучения его наследственного анамнеза, рентгенологических и молекулярно-генетических исследований, биохимического анализа крови. При осмотре в первую очередь обращают на себя внимание дерматологические проявления заболевания – гипертрофия кожи на голове, лице и конечностях, повышенная сальность и связанные с этим нарушения (акне, угревая сыпь), гипергидроз. Определяется деформация пальцев в виде «барабанных палочек», при худобе пациента также можно выявить утолщение длинных трубчатых костей, особенно в области метафизов. Иногда обнаруживаются отечность и болезненность суставов, больные пахидермопериостозом жалуются боли в костях, быструю утомляемость, разбитость. Гистологическое исследование кожи выявляет резкое утолщение всех ее слоев, увеличение количества коллагеновых волокон, размеров сальных и потовых желез.
Рентгенологические исследования могут свидетельствовать о незаращении родничков черепа (у маленьких детей) и утолщении костей по причине периостоза. Поверхность костей редко гладкая, чаще бахромчатая или игольчатая. У взрослых людей изменения и дефекты костей черепа при пахидермопериостозе, как правило, не определяются. Общие и биохимические анализы крови при этом заболевании также не обнаруживают никаких патологических изменений, но специализированное исследование выявляет крайне высокий уровень простагландина Е. Молекулярно-генетическая диагностика пахидермопериостоза производится посредством автоматического секвенирования последовательностей генов HPGD или SLCO2A1 с целью выявления мутаций, возможно пренатальное определение. Обследование сердца (например, ЭхоКГ) может подтвердить наличие такого порока, как открытый Боталлов проток.
Дифференциальная диагностика
Дифференциальную диагностику следует производить с вторичными формами этого состояния, поэтому все больные в обязательном порядке проходят обследование легких – в особенности на предмет злокачественной опухоли.
Лечение
Специфического лечения пахидермопериостоза не существует, применяется симптоматическая терапия, иногда проводятся корректирующие мероприятия. Для уменьшения выраженности симптомов назначают кортикостероиды (как системно, так и местно, в виде мазей или лекарственного электрофореза), препараты, улучшающие трофику тканей. Для уменьшения болезненности суставов и улучшения общего состояния больных пахидермопериостозом используют нестероидные противовоспалительные средства – диклофенак натрия, ибупрофен, индометацин. Они не только обладают анальгезирующим действием, но и угнетают образование простагландинов за счет блокировки фермента циклооксигеназы. Для уменьшения выраженности гипергидроза применяют местные антиперспиранты и такие методики, как ботулинотерапия.
В ряде случаев для устранения проявлений пахидермопериостоза прибегают к хирургическим вмешательствам. В первую очередь это относится к коррекции пороков сердца – для этого производят операцию по закрытию артериального протока. Также могут быть использованы методики пластической хирургии для устранения выраженных эстетических недостатков, обусловленных дерматологическими проявлениями заболевания. Хирургическая ортопедия используется в случае развития контрактур суставов и других выраженных нарушений опорно-двигательного аппарата, являющихся следствием пахидермопериостоза.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: L62,0 Булавовидный ноготь при пахидермопериостозе.
L62.0 Булавовидный ноготь при пахидермопериостозе
Описание
Пахидермопериостоз. Генетически гетерогенное заболевание, которое проявляется комплексным поражением кожных покровов, суставов, костей и ряда внутренних органов. Симптомами этого состояния являются гиперплазия всех слоев кожи, гипергидроз, нарушения со стороны сердечно-сосудистой системы, деформация дистальных фаланг пальцев по типу «барабанных палочек». Диагностика пахидермопериостоза осуществляется на основании данных осмотра пациента, рентгенологических и биохимических исследований, изучения наследственного анамнеза и молекулярно-генетического анализа. Специфического лечения заболевания не существует, но симптоматические мероприятия способны длительное время сдерживать его развитие.
Дополнительные факты
Пахидермопериостоз (первичная гипертрофическая остеоартропатия) – наследственное заболевание с различным (согласно последним открытиям в области генетики) механизмом наследования, поражающее кожные покровы, опорно-двигательный аппарат и сердечно-сосудистую систему. По некоторым данным, этой состояние впервые было описано еще в 1868-м году Г. Фридрайхом, однако его полноценное исследование и определение в качестве отдельной нозологической единицы было осуществлено А. Туреном только в 1935-м году. Основной причиной столь долгой невозможности определить пахидермопериостоз, как самостоятельное заболевание, стало то, что многие его проявления схожи с вторичными (симптоматическими) нарушениями, обусловленными поражением легких и сердца или онкологическими процессами. Данная патология намного чаще поражает мужчин (половое распределение заболевших составляет 8:1) и при этом протекает у них в целом тяжелее, нежели у женщин. Встречаемость пахидермопериостоза в настоящий момент точно не определена – основными препятствиями для этого является частая ошибочная диагностика с симптоматическими формами и наличие стертых вариантов заболевания, наблюдаемое у гетерозигот.
L62.0 Булавовидный ноготь при пахидермопериостозе
Причины
Изучение этиологии и патогенеза пахидермопериостоза продолжалось очень долго, только в 2008 году удалось идентифицировать первый ген, ответственный за развитие этого заболевания. Им является HPGD, расположенный на 4-й хромосоме и кодирующий фермент NAD+-зависимой 15-гидрокси-простогландин дегидрогеназы. Функцией этого белка является дегидратация простагландина Е, процесс считается важным звеном метаболизма и утилизации этого биологически активного вещества. В результате мутации HPGD данный процесс нарушается, концентрация простагландина увеличивается, что и является причиной развития пахидермопериостоза. В настоящий момент среди врачей-генетиков ведется дискуссия по поводу механизма наследования мутаций этого гена – большинство склоняется к тому, что он является аутосомно-рецессивным. Но наличие стертых симптомов пахидермопериостоза у гетерозигот по дефектному гену HPGD дает основания некоторым специалистам заявлять о аутосомно-доминантном наследовании с неполной пенетрантностью. Вариант этой патологии, обусловленный мутацией HPGD, был назван первичным гипертрофической остеоартропатией 1-го типа.
Другая форма пахидермопериостоза была открыта вскоре после идентификации гена HPGD – оказалось, что у некоторых больных с доказанным семейным характером патологии отсутствуют такие генетические дефекты. В итоге была выявлена первичная гипертрофическая остеоартропатия 2-го типа, обусловленная мутацией гена SLCO2A1, локализованного на 3-й хромосоме. Продуктом экспрессии данного гена является трансмембранный белок, облегчающий транспорт молекул простагландинов через биологические мембраны. Нарушение структуры этого протеина, вызванное генетическими дефектами, приводит к ухудшению транспорта простагландинов и их накоплению в кровотоке, что, как и в предыдущем случае, является причиной пахидермопериостоза. Формы этого заболевания, обусловленные мутацией гена SLCO2A1, наследуются по аутосомно-рецессивному механизму.
Классификация
В настоящее время выделяют также малоизученную аутосомно-доминантную разновидность пахидермопериостоза. Она является наиболее редкой, и ее этиология до сих пор неясна – по некоторым данным, это могут быть как варианты мутации HPGD, так и повреждения других, еще не идентифицированных генов. Клинические исследования таких больных (в частности, определение уровня простагландина Е) указывают на то, что патогенез этой формы пахидермопериостоза мало отличается от других типов заболевания, но имеет ярко выраженный аутосомно-доминантный характер наследования.
Симптомы
Пахидермопериостоз характеризуется довольно разнообразной клинической картиной, особенно в отношении первых симптомов заболевания. В большинстве случаев проявления патологии можно обнаружить уже в первые несколько лет жизни ребенка, но наиболее выраженными они становятся к подростковому возрасту. Описаны случаи развития симптомов пахидермопериостоза у взрослых людей, которые до этого были фенотипически здоровыми. В случае раннего начала заболевания одним из первых проявлений будет нарушение и задержка зарастания родничков на черепе, в дальнейшем присоединяются дерматологические симптомы. К ним относят увеличение толщины кожных покровов на лице, голове, кистях и стопах, развитие их складчатости, иногда в сочетании с гиперкератозом. Достаточно сильно при пахидермопериостозе страдают веки – происходит утолщение хрящевой основы и кожи, дистрофия конъюнктивы, развитие кист из тарзальных желез. Характерен также выраженный ладонный и стопный гипергидроз.
Диагностика
Для определения наличия пахидермопериостоза используют данные осмотра больного, изучения его наследственного анамнеза, рентгенологических и молекулярно-генетических исследований, биохимического анализа крови. При осмотре в первую очередь обращают на себя внимание дерматологические проявления заболевания – гипертрофия кожи на голове, лице и конечностях, повышенная сальность и связанные с этим нарушения (акне, угревая сыпь), гипергидроз. Определяется деформация пальцев в виде «барабанных палочек», при худобе пациента также можно выявить утолщение длинных трубчатых костей, особенно в области метафизов. Иногда обнаруживаются отечность и болезненность суставов, больные пахидермопериостозом жалуются боли в костях, быструю утомляемость, разбитость. Гистологическое исследование кожи выявляет резкое утолщение всех ее слоев, увеличение количества коллагеновых волокон, размеров сальных и потовых желез.
Рентгенологические исследования могут свидетельствовать о незаращении родничков черепа (у маленьких детей) и утолщении костей по причине периостоза. Поверхность костей редко гладкая, чаще бахромчатая или игольчатая. У взрослых людей изменения и дефекты костей черепа при пахидермопериостозе, как правило, не определяются. Общие и биохимические анализы крови при этом заболевании также не обнаруживают никаких патологических изменений, но специализированное исследование выявляет крайне высокий уровень простагландина Е. Молекулярно-генетическая диагностика пахидермопериостоза производится посредством автоматического секвенирования последовательностей генов HPGD или SLCO2A1 с целью выявления мутаций, возможно пренатальное определение. Обследование сердца (например, ЭхоКГ) может подтвердить наличие такого порока, как открытый Боталлов проток.
Дифференциальная диагностика
Дифференциальную диагностику следует производить с вторичными формами этого состояния, поэтому все больные в обязательном порядке проходят обследование легких – в особенности на предмет злокачественной опухоли.
Лечение
Специфического лечения пахидермопериостоза не существует, применяется симптоматическая терапия, иногда проводятся корректирующие мероприятия. Для уменьшения выраженности симптомов назначают кортикостероиды (как системно, так и местно, в виде мазей или лекарственного электрофореза), препараты, улучшающие трофику тканей. Для уменьшения болезненности суставов и улучшения общего состояния больных пахидермопериостозом используют нестероидные противовоспалительные средства – диклофенак натрия, ибупрофен, индометацин. Они не только обладают анальгезирующим действием, но и угнетают образование простагландинов за счет блокировки фермента циклооксигеназы. Для уменьшения выраженности гипергидроза применяют местные антиперспиранты и такие методики, как ботулинотерапия.
В ряде случаев для устранения проявлений пахидермопериостоза прибегают к хирургическим вмешательствам. В первую очередь это относится к коррекции пороков сердца – для этого производят операцию по закрытию артериального протока. Также могут быть использованы методики пластической хирургии для устранения выраженных эстетических недостатков, обусловленных дерматологическими проявлениями заболевания. Хирургическая ортопедия используется в случае развития контрактур суставов и других выраженных нарушений опорно-двигательного аппарата, являющихся следствием пахидермопериостоза.
Источник