Особый эпилептический синдром мкб 10

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
G40,5 Особые эпилептические синдромы.
G40.5 Особые эпилептические синдромы
Синонимы диагноза
Особые эпилептические синдромы, синдром веста.
Описание
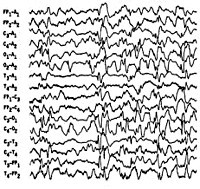
Синдром Веста. Серийные спастические сокращения в отдельных мышечных группах или генерализованного характера, протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ — паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. В большинстве случаев имеет симптоматический характер. Диагностика синдрома основана на клинических данных и результатах ЭЭГ. Для выявления основной патологии необходимы КТ или МРТ, ПЭТ головного мозга, консультация генетика, нейрохирурга. Лечение возможно противоэпилептическими препаратами, стероидами (АКТГ, преднизолон), вигабатрином. По показаниям решается вопрос о хирургическом лечении (каллозотомия, удаление патологического очага).
G40.5 Особые эпилептические синдромы
Дополнительные факты
Синдром Веста носит название по имени врача, наблюдавшего его проявления у своего ребенка и впервые описавшего его в 1841 г. В связи с манифестацией синдрома в раннем возрасте и протеканием судорог по типу серии отдельных спазмов, пароксизмы, характеризующие синдром Веста, получили название инфантильные спазмы. Первоначально заболевание относили к генерализованной эпилепсии. В 1952 г. Был изучен специфический гипсаритмический ЭЭГ-паттерн, патогномоничный для этой формы эпилепсии и характеризующийся медленноволновой асинхронной активностью с беспорядочными спайками высокой амплитуды. В 1964 г. Специалистами в области неврологии синдром Веста был выделен в качестве отдельной нозологии.
Внедрение в неврологическую практику нейровизуализации позволило определить наличие у пациентов очаговых поражений вещества мозга. Это заставило неврологов пересмотреть свои взгляды на синдром Веста как на генерализованную эпилепсию и отнести его в ряд эпилептических энцефалопатий. В 1984 г. Был выявлена эволюция эпилептической формы энцефалопатии от ее раннего варианта в синдром Веста, а с течением времени в синдром Леннокса-Гасто.
В настоящее время синдром Веста занимает около 2% от всех случаев эпилепсии у детей и примерно четверть младенческой эпилепсии. Распространенность составляет, по различным источникам, от 2 до 4,5 случаев на 10 тыс. Новорожденных. Несколько чаще заболевают мальчики (60%). 90% случаев манифестации синдрома приходится на 1-й год жизни, с пиком в возрасте от 4 до 6 мес. Как правило, к возрасту 3-х лет мышечные спазмы проходят или трансформируются в иные формы эпилепсии.
Причины
В подавляющем большинстве случаев синдром Веста носит симптоматический характер. Он может возникать вследствие перенесенных внутриутробных инфекций (цитомегалии, герпетической инфекции), постнатального энцефалита, гипоксии плода, преждевременных родов, внутричерепной родовой травмы, асфиксии новорожденного, постнатальной ишемии вследствие позднего пережатия пуповины. Синдром Веста может являться следствием аномалий строения головного мозга: септальной дисплазии, гемимегалоэнцефалии, агенезии мозолистого тела и пр. В ряде случаев инфантильные спазмы выступают симптомом факоматозов (синдрома недержания пигмента, туберозного склероза, нейрофиброматоза), точечных генных мутаций или хромосомных аберраций (в т. Синдрома Дауна). В литературе упоминаются случаи фенилкетонурии с инфантильными спазмами.
В 9-15% синдром Веста является идиопатическим или криптогенным, т. Е. Его первопричина не установлена или не очевидна. Зачастую при этом прослеживается наличие случаев фибрильных судорог или эпиприступов в семейном анамнезе больного ребенка, т. Е. Имеет место наследственная предрасположенность. Ряд исследователей указывают, что фактором, провоцирующим синдром Веста, может выступать вакцинация, в частности введение АКДС. Это может быть связано с совпадением сроков вакцинации и возраста типичного дебюта синдрома. Однако достоверные данные, подтверждающие провоцирующую роль вакцин, пока не получены.
Патогенетические механизмы возникновения инфантильных спазмов являются предметом изучения. Существует несколько гипотез. Одна из них связывает синдром Веста с расстройством функционирования серотонинергических нейронов. Действительно, у пациентов наблюдается понижение уровня серотонина и его метаболитов. Но пока неизвестно, является оно первичным или вторичным. Обсуждалась также иммунологическая теория, связывающая синдром Веста с увеличением количества активированных В-клеток. Положительный лечебный эффект АКТГ лег в основу гипотезы о сбоях в системе «мозг-надпочечники». Отдельные исследователи предполагают, что в основе синдрома лежит избыточное количество (гиперэкспрессия) возбуждающих синапсов и проводящих коллатералей, формирующих повышенную возбудимость коры. Асинхронность ЭЭГ-паттерна они связывают с физиологичным для этого возрастного периода недостатком миелина. По мере созревания мозга происходит уменьшение его возбудимости и нарастание миелинизации, что объясняет дальнейшее исчезновение пароксизмов или их трансформацию в синдром Леннокса-Гасто.
Симптомы
Как правило, симптом Веста дебютирует на первом году жизни. В отдельных случаях его манифестация происходит в более старшем возрасте, однако не позже 4-х лет. Основу клиники составляют серийные мышечные спазмы и нарушение психомоторного развития. Первые пароксизмы зачастую появляются на фоне уже существующей задержки психомоторного развития (ЗПР), но в 1/3 случаев возникают у первично здоровых детей. Отклонения в нейропсихологическом развитии наиболее часто проявляются снижением и выпадением хватательного рефлекса, аксональной гипотонией. Возможно отсутствие слежения глазами за предметами и расстройство фиксации взора, что является прогностически неблагоприятным критерием.
Мышечные спазмы носят внезапный симметричный и кратковременный характер. Типична их серийность, при этом интервал между следующими друг за другом спазмами длится не менее 1 минуты. Обычно наблюдается возрастание интенсивности спазмов в начале пароксизма и ее спад в конце. Число спазмов, происходящих за сутки, варьирует от единиц до сотен. Наиболее часто возникновение инфантильных спазмов происходит в период засыпания или сразу после сна. Провоцировать пароксизм способны резкие громкие звуки и тактильная стимуляция.
Семиотика пароксизмов, которыми сопровождается синдром Веста, зависит от того, какая мышечная группа сокращается — экстензорная (разгибательная) или флексорная (сгибательная). По этому признаку спазмы классифицируют на экстензорные, флексорные и смешанные. Чаще всего наблюдаются смешанные спазмы, затем сгибательные, наиболее редко — разгибательные. В большинстве случаев у одного ребенка наблюдаются спазмы нескольких видов и то, какой именно спазм будет преобладать, зависит от положения тела в момент начала пароксизма.
Может иметь место генерализованное сокращение всех мышечных групп. Но более часто наблюдаются локальные спазмы. Так, судороги в сгибателях шеи сопровождаются кивками головой, спазмы в мускулатуре плечевого пояса напоминают пожимание плечами. Типичным является пароксизм по типу «складного ножа», обусловленный сокращением мышц сгибателей живота. При этом тело как бы складывается пополам. Инфантильные спазмы верхних конечностей проявляются отведением и приведением рук к туловищу; со стороны кажется, что ребенок сам себя обнимает. Сочетание подобных спазмов с пароксизмом по типу «складного ножа» ассоциируется с принятым на Востоке приветствием «салаам», поэтому было названо «салаамовой атакой». У детей, которые умеют ходить, спазмы могут протекать по типу дроп-атак — неожиданных падений с сохранением сознания.
Диагностика
Синдром Веста диагностируется по основной триаде признаков: приступы кластерных мышечных спазмов, задержка психомоторного развития и гипсаритмический ЭЭГ-паттерн. Имеют значение возраст манифестации спазмов и их связь со сном. Трудности диагностики возникают при позднем дебюте синдрома. В ходе диагностики ребенок консультируется педиатром, детским неврологом, эпилептологом, генетиком.
Дифференциальная диагностика
Дифференцировать синдром Веста следует с доброкачественным младенческим миоклонусом, доброкачественной роландической эпилепсией, младенческой миоклонической эпилепсией, синдромом Сандифера (наклон головы по типу кривошеи, гастроэзофагальный рефлюкс, эпизоды опистотонуса, которые могут быть приняты за спазмы).
Интериктальная (межприступная) ЭЭГ характеризуется наличием дезорганизованной беспорядочной, динамично изменяющейся спайк-волновой активности, как в период бодрствования, так и во сне. Проведение полисомнографии позволяет выявить отсутствие спайк-активности в период глубоких стадий сна. Гипсаритмия регистрируется в 66% случаев, обычно на ранних стадиях. Позже наблюдается некоторая организация хаотичного ЭЭГ-паттерна, а в возрасте 2-4 лет его переход в комплексы «острая-медленная волна». Наиболее частый иктальный ЭЭГ-паттерн (т. Е. ЭЭГ-ритм в период спазмов) — это генерализованные медленноволновые комплексы высокой амплитуды с последующим угнетением активности не менее 1 сек. При регистрации на ЭЭГ фокальных изменений следует думать об очаговом характере поражения головного мозга или наличии аномалий его строения.
КТ головного мозга у имеющих синдром Веста детей может выявлять диффузные либо очаговые изменения церебральных структур, но может быть в пределах нормы. В диагностике локальных поражений более чувствительным методом является МРТ головного мозга. Для выявления участков гипометаболизма мозговых тканей в некоторых случаях возможно проведение ПЭТ головного мозга.
Лечение
Синдром Веста считался резистентным к проводимой терапии вплоть до открытия в 1958 г. Влияния на приступы препаратов АКТГ. Терапия АКТГ и преднизолоном приводит к значительному улучшению или полному прекращению инфантильных спазмов, что сопровождается исчезновением гипсаритмического ЭЭГ-паттерна. До сих пор среди неврологов нет однозначных решений касательно доз и длительности стероидной терапии. Исследования показали, что в 90% случаев терапевтический успех достигался при применении больших дозировок АКТГ. Сроки терапии могут варьировать в пределах 2-6 недель.
Новый этап в лечении инфантильных спазмов начался в 1990-1992 гг. После обнаружения положительного терапевтического эффекта вигабатрина. Однако преимущество лечения вигабатрином пока доказано лишь для больных туберозным склерозом. В остальных случаях исследования показали большую эффективность стероидов. С другой стороны стероидная терапия имеет худшую, в сравнении с вигабатрином, переносимость и более высокий процент рецидивов.
Из антиконвульсантов эффективность показана лишь у нитразепама и вальпроевой кислоты. У отдельных пациентов описан лечебный эффект больших доз витамина В6, который отмечался в первые недели терапии. При инфантильных спазмах, резистентных к проводимой терапии, с подтвержденным на томографии наличием патологического очага показана консультация нейрохирурга для решения вопроса о резекции очага. Если подобная операция невозможна, то при наличии дроп-атак проводится тотальная каллозотомия (пересечение мозолистого тела).
Прогноз
Обычно к 3-летнему возрасту наблюдается регресс и исчезновение инфантильных спазмов. Но примерно в 55-60% случаев они трансформируются в другую форму эпилепсии, чаще всего в синдром Леннокса-Гасто. Фармакорезистентность часто констатируется при инфантильных спазмах, сопровождающих синдром Дауна. Даже при успешном купировании пароксизмов синдром Веста имеет неудовлетворительный прогноз в плане психомоторного развития ребенка. Возможны когнитивные и поведенческие нарушения, ДЦП, аутизм, трудности в обучении. Остаточный психомоторный дефицит не наблюдается только в 5-12% случаев. ЗПР отмечается у 70-78% детей, двигательные расстройства — у 50%. Серьезный прогноз имеет синдром Веста, обусловленный аномалиями или дегенеративными изменениями головного мозга. При этом летальность может достигать 25%.
Более благоприятный прогноз имеют криптогенный и идиопатический синдром Веста при отсутствии ЗПР до появления спазмов. В этой группе больных остаточный интеллектуальный или неврологический дефицит отсутствует у 37-44% детей. Неблагоприятно отражается на прогнозе болезни откладывание начала лечения. Прогностическая оценка затрудняется тем, что отдаленные последствия также зависят от основной патологии, на фоне которой возникает симптоматический синдром Веста.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: G40.5
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G40-G47 Эпизодические и пароксизмальные расстройства / G40 Эпилепсия
Определение и общие сведения[править]
Синдром эпилепсии, индуцированный фебрильной инфекцией
Синонимы: синдром FIRES, острый энцефалит с рефрактерными повторяющимися парциальными припадками, острый негерпетический энцефалит с тяжелым рефрактерным эпилептическим статусом, изнуряющая эпилептическая энцефалопатия у детей школьного возраста, идиопатическая катастрофическая эпилептическая энцефалопатия
Синдром эпилепсии, индуцированный фебрильной инфекцией (FIRES) — это потенциально фатальная острая эпилептическая энцефалопатия с резким началом, которая развивается у ранее здоровых детей и подростков после периода неспецифической лихорадки.
Распространенность синдрома эпилепсии, индуцированный фебрильной инфекцией (FIRES) в составляет 1/100 000 детей и подростков, ежегодная заболеваемость 1/1 000 000 человек.
Этиология и патогенез[править]
В настоящее время этиология неизвестна. Предполагается инфекционный патогенез, но прямые причины заболевания пока не установлены. Возможна также генетическая этиология заболевания, по аналогии с синдромом Драве, но каузальные гены пока также не выявлены. Было доказано, что существует иммунная подоснова для развития синдрома FIRES, поскольку определенные аутоиммунные антитела, такие как VGKC, anti-GAD и anti GluR-3, обнаруживаются у некоторых пациентов. Однако большинство пациентов с синдромом эпилепсии, индуцированной фебрильной инфекцией (FIRES) в основном являются устойчивыми к иммунотерапии и у них не выявляются аутоантитела. Метаболический дефект, например, митохондриальная энцефалопатия, обсуждается как возможный этиологический фактор.
Клинические проявления[править]
Синдром эпилепсии, индуцированный фебрильной инфекцией (FIRES) обычно манифестирует в возрасте 3-15 лет у ранее здоровых детей с нормальными физическим развитием. Начало синдрома FIRES всегда происходит после заболевания, сопровождающегося лихорадкой. Симптомы включают в себя внезапное начало конвульсивных и рецидивирующих фокальных судорожных припадков. За этим следует развитие рефрактерной фокальной эпилепсии со снижением памяти, когнитивных функций и нарушениями поведения. В некоторых случаях могут возникать психические расстройства и иногда моторная недееспособность. В серьезных случаях прогрессирование патологии может привести к вегетативному или полусознательному состоянию или даже смерти.
Особые эпилептические синдромы: Диагностика[править]
Синдром эпилепсии, индуцированный фебрильной инфекцией (FIRES) следует подозревать, когда имеется острое начало рефрактерных судорожных припадков через несколько днейпосле заболевания, сопровождающегося лихорадкой, у ранее здоровых детей. Анализ цереброспинальной жидкости показывает нормальные результаты или мягкий плеоцитоз, но отсутствие патогенов. Тесты на метаболические заболевания обычно выполняются с отрицательными результатами. МРТ обычно не обнаруживает особых аномалий.
Дифференциальный диагноз[править]
Дифференциальный диагноз эпилепсии, индуцированный фебрильной инфекцией (FIRES) включают синдром Драве, синдром Альперса, женскую ограниченную эпилепсию с умственной неполноценностью из-за мутаций PCDH19, инфекционный или лимбический энцефалит (например, с анти-VGKC антителами или антителами к NMDA-рецепторам) и метаболические заболевания (например, биотин-реагирующее заболевание базальных ганглиев, цитруллинемия).
Особые эпилептические синдромы: Лечение[править]
Пациенты с FIRES требуют немедленной госпитализации. Антиэпилептические препараты назначают для купирования судорог, но они часто неэффективны. Высокие дозы фенобарбитала и клобазама, скорее всего, будут эффективными. В тяжелых случаях барбитураты являются единственным методом лечения рефрактерного эпилептического статуса, однако лечение путем индуцирования длительной спазматической комы ассоциируется с худшим когнитивным исходом. Кетогенная диета была полезной в некоторых случаях, особенно если она была рано начата.
Прогноз
Синдром эпилепсии, индуцированный фебрильной инфекцией (FIRES) часто имеет плохой прогноз, но несколько пациентов полностью выздоровели.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Эпилепсия (МКБ10- G40.0) является довольно распространенным психоневрологическим заболеванием с хроническим латентным характером течения. Данное заболевание характеризуется внезапными эпилептическими припадками, при которых у больных нарушаются чувствительные, двигательные, мыслительные и вегетативные функции.
Долгое время эпилепсия считалась неизлечимой болезнью. Однако, благодаря развитию современной медицины, на сегодняшний день это мнение считается ошибочным. Применение противоэпилептических препаратов позволяет преодолеть заболевание более чем в 50% случаев. У остальных больных отмечается существенное уменьшение его клинических проявлений.
: причины развития, симптомы и лечение")
Качественную диагностику и эффективное лечение эпилепсии предлагает клиника неврологии Юсуповской больницы в Москве. В клинике имеется вся необходимая медицинская аппаратура, применяются новейшие методики, гарантирующие постановку точного диагноза и высокие результаты противоэпилептической терапии.
Основными методами борьбы с эпилепсией являются: длительная, регулярная и постоянная медикаментозная терапия, а также соблюдение здорового образа жизни.
Классификация
Согласно МКБ (Международной классификации болезней), существует три этиологических формы эпилепсии:
- идиопатическая эпилепсия (МКБ10 — G40.0) – при отсутствии явной причины;
- криптогенная эпилепсия (МКБ10 — G40.0) – если причина не ясна (не установлена);
- симптоматическая эпилепсия (МКБ10 — G40.2).
Приступы эпилепсии, возникающие у людей среднего и старшего возраста, развиваются, как правило, вследствие различных заболеваний или негативного воздействия внешних факторов. В таких случаях можно подозревать симптоматическую (вторичную) эпилепсию или эписиндром.
Как и любая другая форма данного заболевания, симптоматическая эпилепсия (МКБ 10- G 40.2) может быть генерализованной и локализованной.
Развитие генерализованной симптоматической эпилепсии вызвано изменением глубинных отделов головного мозга и дальнейшим распространением патологического процесса на весь орган.
Локализованная симптоматическая эпилепсия возникает в случае, если поражается какой-либо определенный участок головного мозга и нарушается прохождение сигналов в его коре. Данная форма заболевания, в свою очередь, подразделяется на несколько видов:
- симптоматическую лобную эпилепсию;
- симптоматическую височную эпилепсию;
- симптоматическую эпилепсию затылочной доли;
- симптоматическую эпилепсию теменной доли.
Причины развития
Наиболее часто возникновение симптоматических эпилептических припадков обусловлено следующими провоцирующими факторами:
- объемными образованиями центральной нервной системы;
- наследственными заболеваниями обмена веществ;
- алкогольной абстиненцией;
- инфекционными болезнями нервной системы (например, менингоэнцефалитом);
- черепно-мозговыми травмами (спонтанной или травматической внутричерепной гематомой, ушибами головного мозга);
- длительной бессонницей и психологическим стрессом;
- эклампсией;
- мозговым инфарктом, тромбозом венозного синуса;
- гипервентиляцией, мерцающим светом (при наличии фоточувствительности).
Признаки заболевания
Симптоматическая эпилепсия может проявляться по-разному у пациентов с различными формами данного заболевания.
При генерализованных приступах больные, как правило, теряют сознание, полностью утрачивают контроль над своими действиями, могут упасть. У них развивается ярко выраженный судорожный синдром.
Симптомы локализованной эпилепсии могут быть разнообразными и зависят от локализации очага поражения. Они бывают психическими, чувственными, вегетативными или двигательными.
По степени тяжести симптоматическая эпилепсия может быть легкой или тяжелой.
Легкие приступы обычно не сопровождаются потерей сознания, однако у больных наблюдается возникновение обманных, непривычных ощущений, они могут терять контроль над частями тела.
При тяжелой симптоматической эпилепсии человек может потерять связь с реальностью, у него судорожно сокращаются определенные группы мышц, утрачивается контроль над собственными движениями.
Лобная симптоматическая эпилепсия проявляется внезапным началом приступа, его короткой протяженностью (от 40 до 60 секунд) и высокой частотой, двигательными феноменами.
У больных с височной симптоматической эпилепсией наблюдается возникновение спутанности сознания, слуховых и зрительных галлюцинаций, лицевых и кистевых автоматизмов.
При теменной симптоматической эпилепсии у пациентов происходит развитие мышечных спазмов, болевых ощущений, приступов сексуального желания, нарушается температурное восприятие.
Затылочная симптоматическая эпилепсия характеризуется появлением зрительных галлюцинаций, неконтролируемым морганием, нарушения поля зрения, подергиванием головы.
Диагностика и лечение в Юсуповской больнице
Симптоматическая эпилепсия может диагностироваться в случае неоднократного повторения приступов. Диагностика эпилепсии в клинике неврологии Юсуповской больницы представляет собой комплексное обследование, состоящее из нескольких этапов:
- развернутого клинического и биохимического анализов крови, анализа мочи – позволяющих определить признаки инфекции, электролитный баланс, анемию или сахарный диабет, которые могут провоцировать припадки;
- электроэнцефалограммы (ЭЭГ) – является наиболее распространенным тестом для выявления эпилепсии, позволяющим регистрировать общие изменения мозговых волн у больных даже вне припадка;
- позитронно-эмиссионной томографии (ПЭТ);
- обзорной и контрастной рентгенографии;
- ревизии диска МРТ (магнитно-резонансной томографии).
Применение самых передовых диагностических методик и новейшая медицинская аппаратура клиники неврологии Юсуповской больницы гарантируют оперативную постановку правильного диагноза.
Лечение симптоматической эпилепсии зависит от основной патологии, которая привела к ее развитию. Как правило, симптоматическая эпилепсия хорошо поддается терапии, направленной на прекращение эпилептических припадков и многим пациентам Юсуповской больницы, уже удалось убедиться в успешности назначенного лечения.
Для каждого больного в Юсуповской больнице разрабатывается индивидуальная, комплексная терапевтическая схема, может быть назначено проведение медикаментозной терапии с использованием современных препаратов, безоперационной электрической стимуляции головного мозга через блуждающий нерв либо эмболизации сосудов головного мозга.
Благодаря современному лечебно-диагностическому оборудованию клиники, большому практическому опыту и высокой компетенции врачей эпилептологов Юсуповской больницы обеспечиваются максимально высокие результаты даже в самых тяжелых случаях эпилепсии.
Практически во всех случаях помогает медикаментозное лечение, лишь некоторым пациентам требуется проведение хирургического лечения, позволяющего устранить патологию.
Грамотная терапия, которую подбирают высококвалифицированные эпилептологи Юсуповской больницы, способствует сведению возможных побочных эффектов к минимуму, после чего становится возможной последующая постепенная отмена приема лекарственных препаратов без возобновления эпилептических припадков.
Записаться на прием к врачу-эпилептологу, получить информацию о стоимости услуг по диагностике и лечению эпилепсии, условиях госпитализации больного можно по телефону Юсуповской больницы или на сайте клиники, связавшись с нашими врачами-координаторами.
Автор

Эпилептолог, кандидат медицинских наук
Список литературы
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Брюханова Н.О., Жилина С.С., Айвазян С.О., Ананьева Т.В., Беленикин М.С., Кожанова Т.В., Мещерякова Т.И., Зинченко Р.А., Мутовин Г.Р., Заваденко Н.Н.. Синдром Айкарди–Гутьерес у детей с идиопатической эпилепсией // Российский вестник перинатологии и педиатрии. — 2016. — № 2. — С. 68–75.
- Виктор М., Роппер А. Х. Руководство по неврологии по Адамсу и Виктору : учеб. пособие для системы послевуз. проф. образования врачей / Морис Виктор, Аллан Х. Роппер; науч. ред. В. А. Парфенов; пер. с англ. под ред. Н. Н. Яхно. — 7-е изд. — М.: Мед. информ. агентство, 2006. — 677 с.
- Розенбах П. Я.,. Эпилепсия // Энциклопедический словарь Брокгауза и Ефрона : в 86 т. (82 т. и 4 доп.). — СПб., 1890—1907.
Наши специалисты

Невролог, кандидат медицинских наук

Невролог, кандидат медицинских наук

Заведующий отделением восстановительной медицины, врач по лечебной физкультуре, невролог, рефлексотерапевт

Невролог

Невролог

Невролог, ведущий специалист отделения неврологии
Невролог
Цены на услуги *
*Информация на сайте носит исключительно ознакомительный характер. Все материалы и цены, размещенные на сайте, не являются публичной офертой, определяемой положениями ст. 437 ГК РФ. Для получения точной информации обратитесь к сотрудникам клиники или посетите нашу клинику.
Скачать прайс на услуги
Мы работаем круглосуточно
Источник