Операция на аорте с марфан синдром

Разрывы и расслоение стенки аорты остаются актуальной проблемой. Они являются основной и самой частой причиной смерти у больных синдромом Марфана (СМ).
Прогрессирующая эктазия восходящей аорты у больных СМ, приводит к формированию аневризмы аорты, фатальному разрыву аорты или расслоению ее стенки. Ранняя диагностика и своевременное лечение сосудистых осложнений у этих больных является чрезвычайно важным процессом и значительно улучшает продолжительность жизни (Pratt B., Curci J., 2010).
Синдром Марфана — это генетическое заболевание, наследуемое по аутосомно-доминантному типу с характерным поражением сердечно-сосудистой системы, скелета и глаз. В основе СМ лежит дефект гена фибриллина-1 (fibrillin 1, FBN1), который кодирует белок внеклеточного матрикса, входящий в состав эластичных волокон.
Недостаточность FBN1 у мышей является общепринятой моделью СМ, который повторяет сердечно-сосудистый фенотип и показывает дегенеративные изменения в среднем слое стенки аорты, иногда с возникновением воспалительных инфильтратов, ее расширением или расслоением. Второй ген СМ определен недавно как трансформирующий фактор роста β (TGF-β) (Ali R Keramati, 2010; Yu Wang, 2010).
Эластичные волокна — один из важнейших элементов стенки аорты. Механическая или функциональная недостаточность эластина в стенке аорты способствует формированию аневризмы и расслоению стенки.
Роль патологии эластина и эластичных волокон в расслоении аорты (РА) имеет важное значение, особенно у пациентов с СМ. Доказано, что повышение уровня (TGF-β) способствует склонности стенки аорты к расслоению. (На рисунку слева изображены основные типы аневризм). Препараты, которые подавляют TGF-β могут быть полезными для сокращения риска РА у пациентов с СМ (Pratt B., Curci J., 2010).
Заболевания, ассоциированные с аневризмой грудного отдела аорты и расслоением стенки, называли кистозным медиа некрозом, но этот термин является неточным, поскольку болезнь не является связанной с некрозом аорты или формированием кист.
Точный термин «медиа дегенерация» характеризуется триадой изменений:
— потерей гладкомышечных клеток (как правило, участка, а не полной потерей),
— фрагментацией или потерей эластичных волокон
— и увеличением протеогликанов.
Хотя термин «медиа дегенерация» был описан, как невоспалительное заболевания, современные исследователи указывают на наличие воспалительной инфильтрации клеток (ACCF / AHA / AATS / ACR / ASA / SCA / SCAI / SIR / STS / SVM Guidelines, 2010).
Установлено, что воспаление в стенке аорты играет важную роль в прогрессировании расслоения аорты у больных СМ. У больных находят Т-лимфоциты, В-лимфоциты и макрофаги в медии и адвентиции аорты (особенно вокруг vasa vasorum). Существует сходство воспалительных изменений в стенке аорты при СМ и гигантоклеточном артериите и артериите Такаясу. Патогистологические исследования показывают, что Т-лимфоциты проникают в стенку аорты через активацию эндотелиальных клеток в vasa vasorum, а не через эндотелий интимы аорты. Установлено, что иммунный медиа аортит может участвовать в патогенезе аневризмы восходящего отдела аорты у больных СМ, что связано с воспалительным инфильтратом в среднем слое аорты (Rumin He. Et al., 2008).
У больных СМ с аневризмой аорты наблюдаются локальное или глобальное отражение пульсовой волны в аорте. Для их измерения используют эхокардиографию и магнитно-резонансную томографию с контрастированием, чтобы определить центральное давление, тип течения волны, локальный и глобальный коэффициенты отражения. Раннее возвращение отраженных волн повышает систолическое давление и создает дополнительную нагрузку на сердце и крупные сосуды.
Эти волны отражения рассматривают как один из важных факторов, определяющих центральное артериальное давление и как такой, что может способствовать развитию дилатации аорты и ее разрыву. Увеличение длины аорты, с одной стороны и увеличение жесткости стенки аорты с другой способствуют усилению волн отражения. Исследовано связь между тяжестью сердечно-сосудистого фенотипа у больных СМ и типом FBN1 мутации. Также существует корреляция между показателями жесткости стенки аорты и типом FBN1 мутации, но FBN1 генотип не единственный фактор, определяющий жесткость стенки аорты (De Backer J., 2009; Segers P., et al., 2006).
Поражение аорты при СМ регистрируют в 65-100% случаев. В значительном количестве случаев (6-9%) РА, встречается в молодом возрасте с локализацией расслоения в проксимальном отделе аорты. Наследственный дефект соединительной ткани приводит к поражению сердечно-сосудистой системы, а именно – к:
— дилатации синусов Вальсальвы,
— аортальной регургитации,
— аневризме аорты,
— пролапсу митрального клапана (Siepe M., 2009).
Чаще расширяется участок синуса Вальсальвы (56,9%) или восходящей части аорты (63,6%), причем даже значительная дилатация протекает бессимптомно. Иногда отмечают боль за грудиной при физической нагрузке, которая связана с рефлексом перерастянутой аортальной стенки. Достоверная диагностика дилатации синуса Вальсальвы возможна при проведении эхокардиографии.
Рентгенологическое исследование малоинформативное при деформации грудной клетки, которая приводит к изменению позиции аорты и тени сердца относительно позвоночника. По данным Осовской Н.Ю. (2009) у молодых пациентов (15-25 лет) с СМ ширина кольца аорты была достоверно выше, чем в группе контроля, а также втрое чаще наблюдалась аневризма синусов Вальсальвы.
Аневризма синуса Вальсальвы до прорыва не вызывает гемодинамических нарушений и обычно является случайной находкой при эхокардиографическом исследовании. Некоронарный синус повреждается в 26% случаев и обычно прорывается в правое предсердие. Левый коронарный синус повреждается в 5% случаев. Эхокардиография позволяет визуализировать выбухание одного или нескольких синусов в парастернальной проекции по короткой оси на уровне аортального клапана и турбулентный поток крови в той камере, в которой произошел прорыв аневризмы (Martin G., 2008; Осовская Н. Ю. 2008; Siepe M., 2009 ).
Вместе с аневризмой синуса Вальсальвы при синдроме Марфана могут наблюдаться аневризмы восходящей части аорты, дуги, реже — нисходящей части аорты и брюшного отдела аорты. Поражение именно аорты при синдроме Марфана определяет прогноз, и тяжесть заболевания, у основной части больных. Среди взрослых пациентов прогноз для жизни является неблагоприятным.
Смерть наступает у половины больных с СМ среди лиц мужского пола до 40-летнего возраста, а среди женщин — до 50-летнего.
Критические периоды жизни, при которых существует высокий риск разрыва аневризмы — это возраст от 15 до 18 лет и от 40 до 45 лет. Около половины РА встречается у беременных старше 40 лет: чаще — в третьем триместре, редко — в ранний послеродовой период. Причины развития РА по этим показаниям до конца не выяснены, значение придают увеличению объема крови, сердечного выброса и повышению артериального давления. Особенно высок риск РА у женщин с СМ во время беременности. Иногда диагноз СМ устанавливают после диагностики РА в послеродовом периоде (Осовская Н. Ю. 2009; Зильбер А.П., 1999).
Классические рекомендации по лечению СМ предусматривают:
— изменения образа жизни, включая умеренные ограничения физической активности,
— профилактику эндокардита;
— регулярную визуализацию аорты;
— прием медикаментов;
— профилактическую замену восходящей аорты.
На сегодня нет больших, рандомизированных контролируемых клинических исследований по применению медикаментозной терапии у больных СМ. Несмотря на это, обычно, используют четыре группы препаратов, чтобы задержать или заняться профилактикой РА:
— β-блокаторы,
— антагонисты кальция,
— ингибиторы АПФ,
— блокаторы рецепторов ангиотензина II (Stuart Alan Graham, 2007).
Результаты клинических наблюдений показывают, что ранняя хирургическая операция на аорте в случае РА значительно продлевает жизнь больным и служит профилактикой осложнений. В табл. 1 приводим Европейские рекомендации по оперативному лечению больных РА
Таблица 1
Требования к оперативному вмешательству по поводу РА у больных СМ, ESC Guidelines 2010
Показатели | Класса | Уровеньb |
Пациенты должныбытьпрооперированы, еслимаксимальный диаметр корня аорты: | ||
• > 50 мм | I | Cc |
• 46-50 мм если есть: | ||
— Семейный анамнез расслоение аорты или | I | C |
— Прогрессивная дилатация аорты> 2 мм год, которая | I | C |
— Тяжелая аортальная или митральная регургитация или | I | C |
— Желание забеременеть | I | C |
•Показанием коперацииявляется тот факт, если другиечастиаорты >50 мм или дилатация прогрессирует | IIa | C |
a-классрекомендации | ||
Популяционные изменения аортального клапана и восходящего отдела аорты являются стабильными с низким риском и продолжительной работой после оперативного вмешательства (оперативная смертность 1,5% во время плановых операций и 11,7% — во время ургентных). Пяти и десятилетняя выживаемость составляет 84% и 75% соответственно. При синдроме Марфана риск повторного расслоения и рецидивирующих аневризм значительно выше, чем в случае с болезнями аорты другой этиологии.
У пациентов с анатомически нормальными клапанами, у которых недостаточность связана с расширением кольца или расслоением, клапан-сберегающие операции с заменой корня аорты на протез Dacron с реимплантация коронарных артерий в протез (методика David’s) или реконструкция корня аорты (методика Yacoub’s) теперь стали лучшими хирургическими процедурами. Аортальная регургитация является наиболее частым осложнением, которое требует повторной операции у 20% пациентов после десяти лет. Долгосрочные результаты клапан-сохраняющих операций по замене корня аорты при синдроме Марфана все еще неизвестны.
Основным оперативным вмешательством, которое применяется сегодня, является операция, впервые описана в 1968 году двумя хирургами Hugh Bentall и A. De Bono (замена аортального клапана, синуса аорты и восходящей аорты). Искусственные клапаны сердца могут изнашиваться или требуют антикоагулянтов. Часто у больных с СМ обнаруживают нормальный аортальный клапан, что является показанием к клапано-сохраняющей операций. Есть несколько сообщений о краткосрочном успехе после эндоваскулярного стентирования грудной части нисходящей аорты. Установка стента у больных синдромом Марфана не рекомендуется, поскольку риск обычного открытого хирургического вмешательства является минимальным (ESC Guidelines, 2010).Таким образом, разрывы и расслоения стенки аорты у больных СМ остаются актуальной проблемой современной кардиологии и кардиохирургии. Поиск новых методов диагностики и лечения является основной задачей, понимание которой будет способствовать лучшему пониманию механизмов возникновения разрывов и расслоений и позволит врачам проводить профилактику этих состояний у больных СМ.
Литература
1. Зильбер А.П., Шифман Е.М., Егорова И.М., Кузьмина Л.А. (1999) Синдром Марфана и беременность: аспекты интенсивной терапии, анестезии и реанимации. Вестник интенсивной терапии, 2:19-22. 2. Кравченко І.М., Сітар Л.Л., Федонюк Л.Я., Захарова В.П. (2007) Аневризми висхідної аорти та аортальна недостатність при синдромі Марфана: проблеми хірур-гічного лікування та морфології. Клінічна анатомія та оперативна хірургія, 4: 58-61. 3. Кузик Ю.І. (2008) Розшаровуючі аневризми аорти та гостра коронарна недостатність: особливості диференційної діагностики. Український кардіологічний журнал, тези ІX Міжнародного конгресу кардіологів, 2:102-107. 4. Осовська Н. Ю. (2008) Аналіз структурно-функціональних показників серця у пацієнтів з малими структурними серцевими аномаліями. Укр. мед. часопис, 4:54-58. 6. Ситар Л.Л., Кравченко И.Н., Антощенко А.А. и др. (2002) Диагностика и хирургическое лечение травматической аневризмы грудной части аорты. Укр. кардіол. журн., 3:51-54. 7. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. (2010) Circulation, 121: 266-369. 8. De Backer J. (2009) Cardiovascular characteristics in Marfan syndrome and their relation to the genotype. Verhandelingen — Koninklijke Academie voor Geneeskunde van België, 71(6): 335-371. 9. R. Erbel (Chairman), F. Alfonso, C. Boileau et al. (2001) Diagnosis and management of aortic dissection (Recommendations of the Task Force on Aortic Dissection, European Society of Cardiology). European Heart Journal, 22: 1642–1681. 10. Ernst Weigang, Christoph A Nienaber, Tim C Rehders et al. (2008) Management of Patients With Aortic Dissection. Dtsch Arztebl Int, 105: 639-645. 11. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010) (2010). European Heart Journal, 10: 19-20. 12. John Pepper, John Chan, Jemyrr Gavino, et al. (2010) External aortic root support for Marfan syndrome: early clinical results in the first 20 recipients with a bespoke implant. Journal of the Royal Society of Medicine, 103: 370-375. 13. Isselbacher EM, Eagle KA, Zipes DP, et al. (1997). Diseases of the aorta. In Braunwald E. Heart disease: a textbook of cardiovascular medicine (5th ed.). Philadelphia: WB Saunders. pp. 1546–1581. 14. Keramati Ali R, Sadeghpour Anita, Farahani Maryam M, et al. (2010) The non-syndromic familial thoracic aortic aneurysms and dissections maps to 15q21 locus. BMC Med Genet., 11: 143. 15. Klein D.G. (2005) Thoracic aortic aneurysms. J. Cardiovasc. Nurs., 20(4):245-250. 16. Martin G. Keane, Reed E. Pyeritz. (2008) Medical management of Marfan syndrome. Circulation, 117: 2802-2813. T Paul Tran, Ali Khoynezhad (2009) Current management of type B aortic dissection. Vasc Health Risk Manag., 5: 53-63. 17. Pratt B, Curci J. (2010) Arterial elastic fiber structure. Function and potential roles in acute aortic dissection. The Journal of cardiovascular surgery, 51(5): 647-656. 18. Rakesh K Sharma, Donald J Voelker, Rajiv K Sharma, et al. (2010) Coronary computed tomographic angiography (CCTA) in community hospitals: “current and emerging role. Vasc Health Risk Manag., 6: 307-316. 19. Rumin He, Dong-Chuan Guo, Wei Sun, et al. (2008) Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms and sporadic aneurysms. J. Thorac. Cardiovasc. Surg., 136(4): 922-929. 20. Segers P., De Backer J., Devos D. et al. (2006) Aortic reflection coefficients and their association with global indexes of wave reflection in healthy controls and patients with Marfan’s syndrome. American Journal of Physiology — Heart and Circulatory Physiology, 290(6): 2385-2392. 21. Siepe M., Loffelbein F. (2009) The Marfan syndrome and related connective tissue disorders. Med. Monatsschr. Pharm., 32:213. 22. Stuart Alan Graham, Williams Andrew (2007) Marfan’s syndrome and the heart. Archives of Disease in Childhood, 92(4): 351-356. 23. Suziku Toru, Hirohisa Katoh, Ryozo Nagai (1999). Biochemical diagnosis of aortic dissection: from bench to bedside. Japanese Heart Journal, 40(5): 527-534. 24. von Kodolitsch Y, Nienaber C, Dieckmann C, et al. (2004). Chest radiography for the diagnosis of acute aortic syndrome. Am J Med., 116(2): 73-77. 25. Yu Wang, Hafid Ait-Oufella, Olivier Herbin, et al. (2010) TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II–infused mice. J Clin Invest., 120(2): 422–432. |
Автор: Жураев Р.К.
Источник
Синдром Марфана – аутосомно-доминантное наследственное заболевание, обусловленное мутацией гена, кодирующего синтез одного из базовых компонентов соединительной ткани – гликопротеида фибриллина. В частности, генетическое исследование при синдроме выявляет дефект гена FBN1, ответственного за синтез фибриллина-1 и входящего в состав коллагена. Следствием является замена в молекуле аминокислоты пролина на аргинин, что ведет к диспропорциональному превалированию в соединительной ткани коллагена III типа и характерным изменениям ее каркасно-эластических свойств в виде гиперрастяжимости.
Синдром имеет плейотропный характер с одинаковой распространенностью во всех этнических группах (1 случай на 5000) и классическим менделевским наследованием от больного родителя в большинстве случаев, однако у 25-30% пациентов возникает в виде первичной мутации.1 Все многообразие клинических проявлений непосредственно связано с системным вовлечением соединительной ткани. Главную опасность для жизни представляют собой изменения грудной аорты с неизбежным возникновением аневризм и расслоений, непосредственно определяющих прогноз течения заболевания.
Прогноз заболевания до появления хирургических методов лечения аневризм грудной аорты был неблагоприятным, и средняя продолжительность жизни пациентов с синдромом Марфана не превышала 30–40 лет. Летальные исходы в основном обусловлены острым расслоением и разрывом аневризм грудной аорты и/или развитием застойной сердечной недостаточности. В настоящее время в странах с развитым здравоохранением пациенты доживают до преклонного возраста.5
Клинические проявления синдрома Марфана в манифестированных случаях довольно характерны и включают в себя:
- Скелетные аномалии: высокий рост, астеническое телосложение, долихостеномелия (непропорционально длинные руки и ноги), арахнодактилия, воронкообразная или килевидная деформация грудной клетки, кифосколиоз, плоскостопие, долихоцефалия, узкий лицевой скелет («птичье» лицо), «готическое» нёбо. Характерна гиперподвижность суставов. Мышцы часто отстают в росте от скелета, развиты слабо.
- Поражение сердечно–сосудистой системы: патогномонично расширение восходящей аорты, острое расслоение грудной аорты и пролапс митрального клапана.
- Патология зрения: чаще наблюдается миопия вследствие увеличения длины глазного яблока, но возможна гиперметропия. Слабость связочного аппарата хрусталика приводит к его подвывиху или полному вывиху (дислокация хрусталика), что может сопровождаться дрожанием радужной оболочки (иридодонез). Развиваются вторичная глаукома, отслойка сетчатки, катаракта. Характерны голубые склеры.
- Иные полиморфные клинические проявления в виде эмфиземы и спонтанного пневмоторакса за счет разрыва легочных «булл» (чаще у взрослых), гастроптоз, дискинезия желудочно–кишечного тракта, нефроптоз, бедренные, паховые и диафрагмальные грыжи, гипоплазия мышц и подкожной клетчатки, мышечная гипотония и др.
- Психоневрологические нарушения в виде повышенной нервной возбудимости, астено–невротического синдрома, эмоционально-волевых нарушений.
Диагностика
Основывается на Гентских критериях 2010 г. и подтверждается углубленным генетическим анализом с обнаружением дефекта гена FBN1.3
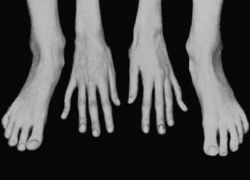
Рис. 1 Характерные проявления синдрома Марфана: длинные и тонкие пальцы (арахнодактилия)
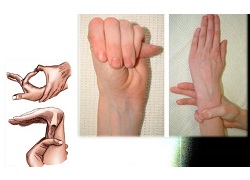
Рис. 2 Диагностические тесты (тест запястья, тест большого пальца)
Клинический случай
Пациентка Б. 1998 года рождения (21 год) поступила в отделение кардиохирургии НМИЦ хирургии им. А.В. Вишневского 31 мая 2019 года. Активно жалоб при поступлении не предъявляет, при детальном расспросе – жалобы на боли в грудной клетке неясной локализации, четко не связанные с физической активностью, одышку, слабость. Из семейного анамнеза известно, что мать пациентки умерла в возрасте 30 лет от острого расслоения аорты. Пациентка с 6 лет была на учете у педиатра с подозрением на синдром Марфана в связи с ранним развитием симптомов данного заболевания: астеническое телосложение, сколиоз грудного отдела позвоночника. С 14 лет находится на учете у кардиолога по поводу диагностированного расширения восходящего отдела аорты. В 15–летнем возрасте генетически подтвержден синдром Марфана, выявлен дефект гена FBN1. С 19–летнего возраста отмечается снижение остроты зрения, миопия слева (–0,75), справа (–1,5).
Ежегодно проводился контроль посредством трансторакальной эхокардиографии и мультиспиральной компьютерной томографии с целью оценки размеров и состояния корня и грудного отдела аорты. До 20–летнего возраста диаметр корня и грудного отдела стабильный, значительного роста этих показателей не отмечается. С 20–летнего возраста наблюдается отрицательная динамика с прогрессивным увеличением размеров восходящего отдела аорты до 44 мм. В 2019 году отмечено резкое увеличение данного сегмента аорты с 45 мм до 50 мм. Дополнительно при МСКТ исследовании в июне 2019 года выявлена локальная диссекция некоронарного синуса аорты.
Также из анамнеза известно, что пациентка в 14–летнем возрасте перенесла субтотальную резекцию щитовидной железы по поводу диффузно–токсического зоба и находится на заместительной терапии L–тироксином 100 мкг.
При осмотре: телосложение астеническое, рост 178 см, вес 65 кг, индекс массы тела 21, имеются лицевые дизморфии (долихоцефалия, гипоплазия скуловых костей, энофтальм), подкожная жировая клетчатка развита слабо. Выявляется левосторонний грудопоясничный сколиоз, умеренная воронкообразная деформация грудной клетки. Отношение длины верхней половины туловища к нижней 0,80, размаха рук к росту 1,17. При аускультации выслушивается диастолический шум во II точке.
Эхокардиографическое исследование выявило умеренную недостаточность аортального клапана 2–2,5 степени, ширина струи регургитации при этом 3,5 мм. Структура створок аортального клапана сохранена, максимальный градиент 4 мм рт.ст. Корень аорты расширен до 38 мм. Митральный и трикуспидальный клапан в пределах нормы. Размеры полостей сердца и насосная функция в пределах нормы, имеется умеренная гипертрофия левого желудочка.
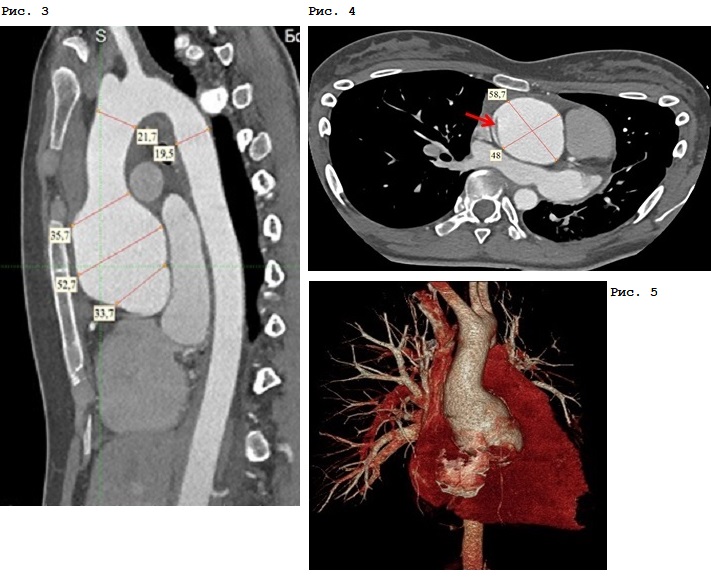
По данным МСКТ грудного отдела аорты с контрастированием наблюдается расширение фиброзного кольца аортального клапана до 32 мм, и тотчас, начиная с уровня синусов Вальсальвы, отмечается расширение аорты до 53 мм, в области синотубулярного соединения до 58х48 мм (рис. 3, красными линиями указаны линейные размеры корня и восходящего отдела аорты). При контрольном исследовании от 03.06.19 по правой и левой полуокружностям восходящего отдела аорты на 53 мм и 32 мм дистальнее фиброзного кольца определяются участки расслоения аорты (рис. 4, красными линиями указаны размеры восходящего отдела аорты; красной стрелкой – зона диссекции). Дуга аорты размерами до 22 мм, не изменена, нисходящий отдел грудной аорты – до 20 мм, также без патологических изменений (рис. 5, 3D–реконструкция грудного отдела аорты).
Диагноз синдрома Марфана был очевиден на основании Гентских критериев, включая семейный анамнез, наличие признаков системного вовлечения соединительной ткани (воронкообразная деформация грудной клетки, кифосколиоз, лицевые дизморфии), наличие аневризмы грудной аорты.
Согласно Рекомендациям по диагностике и лечению заболеваний аорты Европейского общества кардиологов (ESC) от 2014 г., показания к хирургическому вмешательству должны основываться как на размерах аорты, так и особенностях течения заболевания, и риск операции не должен превышать риска его естественного течения. Показания к операции при синдроме Марфана возникают при размерах аорты ≥50 мм. В случае размеров аорты 45–50 мм рассматривается наличие дополнительных факторов риска, таких как семейный анамнез расслоения аорты, быстрый рост диаметра аорты > 3 мм в год и значимая регургитация на аортальном клапане.
Показания к операции в данном клиническом наблюдении не вызывали сомнений, поскольку риск разрыва и расслоения аорты был крайне высокий. Выбор же самой стратегии вмешательства носил дискутабельный характер. Возможно было применить операцию протезирования корня аорты с полным замещением искусственным клапан–содержащим кондуитом или реконструктивную клапан–сохраняющую операцию – с возможностью реимплантации нативного аортального клапана или ремоделирования корня аорты с сохранением аортального клапана.
Операция Бенталла–Де Боно (Bentall–De Bono), или полное замещение корня и восходящего отдела аорты клапан–содержащим кондуитом (рис. 6), – хорошо зарекомендовавшее себя, надежное вмешательство в аортальной хирургии. Имеет ряд очевидных преимуществ, таких как гарантированное длительное функционирование механического протеза и меньшая сложность техники операции. К негативным факторам можно отнести необходимость пожизненного приема антикоагулянтов с постоянным контролем антикоагулянтной терапии. Любая отмена терапии может привести в дисфункции протеза с тяжелыми последствиями. Соответственно, отсутствует возможность беременности и родов.
У молодых женщин с необходимостью сохранения детородной функции возможно применение стратегии клапан–сохраняющих операций в двух вариантах: ремоделирование корня аорты, или операция Якуба (Yacoob) (рис. 7) и реимплантация аортального клапана, или операция Дэвида (David) (рис. 8) в ее модификациях.
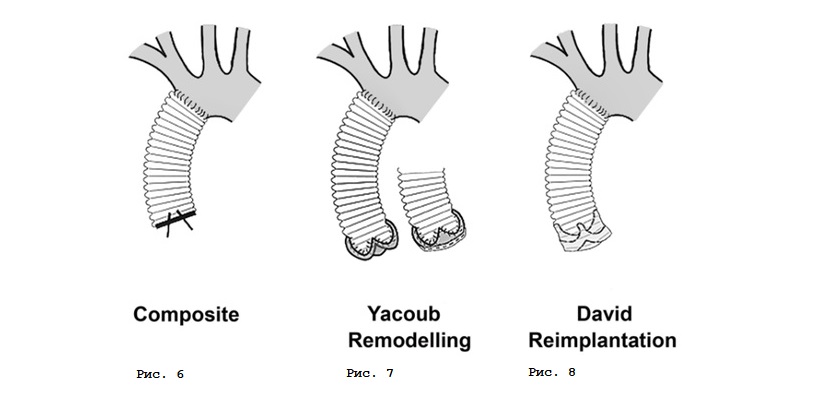
Суть операция Якуба заключается в том, что аортальные синусы заменяются специально смоделированным в виде короны сосудистым протезом таким образом, что сохраняется функциональная связь корня аорты и створок аортального клапана. Второй вариант, более безопасный и воспроизводимый, – операция Дэвида, существующая в нескольких модификациях (I, II, III, IV, V). На сегодняшний день чаще всего используется модификация David V. Суть – в имплантации всех структур аортального клапана внутрь сосудистого протеза с сохранением анатомо–функционального взаимоотношения комиссур и створок аортального клапана. Методику операции отличает надежность гемостаза и функции аортального клапана.
Основным преимуществом обеих клапан-сохраняющих процедур является сохранность нативного клапана, что обеспечивает его хорошую функцию и позволяет отказаться от приема антикоагулянтов.
В нашем случае теоретически имелся риск дисфункции аортального клапана вследствие возможной его некомпетенции в отдаленном периоде по причине системной неполноценности соединительной ткани, в том числе структур самого клапана.
Дискуссия о возможности клапан–сохраняющих операций при синдроме Марфана подошла к завершению только в последние годы после публикации нескольких крупных исследований. В частности, A. Martens с соавт. опубликовали данные 20–летнего наблюдения более сотни пациентов с синдромом Марфана, которым была выполнена процедура David. Авторы пришли к выводу, что клапан–сохраняющие процедуры на корне аорты должны быть рассмотрены именно в качестве предпочтительного метода лечения аневризмы корня аорты у пациентов с синдромом Марфана при сохранных створках клапана. При этом, по–видимому, соединительно–тканная дисфункция не затрагивает структуры аортального клапана и таким образом не имеет негативного влияния на отдаленные результаты вмешательства. Долговечность клапан–сохраняющих процедур при синдроме Марфана обусловлена исключительно правильным отбором пациентов и качеством самой операции. Этим авторы объясняют разнородность полученных ранее результатов в других исследованиях и настаивают на необходимости строгой индивидуальной оценки клапана и достаточного опыта данного вмешательства.8
Еще одно исследование, сравнивающее результаты операций David и Bentall–De Bono у пациентов с синдромом Марфана, опубликовано J. Price с соавт. В частности, сравниваются клапан–сохраняющие процедуры у 98 больных с операцией Bentall–De Bono – у 67. В отдаленном периоде в группе операций Bentall чаще возникали расслоения аорты (25.4 vs 4.1%), умеренная или тяжелая аортальная недостаточность (49.3% vs 14.4%), а также чаще регистрировались экстренные оперативные вмешательства (24.6% vs 3.3%). Госпитальной летальности не было, в отдаленном периоде в сроки до 17 зафиксировано 9 летальных исходов. Соответственно 10–летняя выживаемость в группе Bentall была 90.5%, а в группе клапан–сохраняющих вмешательств – 96.3%. Кроме того, клапан–сохраняющие вмешательства были ассоциированы с меньшим риском тромбоэмболических и геморрагических осложнений, но отдаленная выживаемость и свобода от повторных операций, а также риск вторичного эндокардита в обеих группах были сопоставимы.9
Учитывая вышеперечисленное, нами была выбрана методика клапан–сохраняющей процедуры (табл. 1).
Таблица 1. Преимущества и недостатки стратегий операций вмешательств.
Клапан-сохраняющие процедуры | Протезирование корня аорты | |
Преимущества |
|
|
Недостатки |
|
|
17 июня 2019 г. у данной пациентки нами была выполнена операция Дэвида. Доступ: срединная стернотомия. Канюляция аорты в средней трети дуги аорты канюлей диаметром 21 Fr с использованием методики Сельдингера. Раздельная канюляция ВПВ канюлей 24 Fr. Нижняя полая вена канюлирована через ушко правого предсердия канюлей 32 Fr.
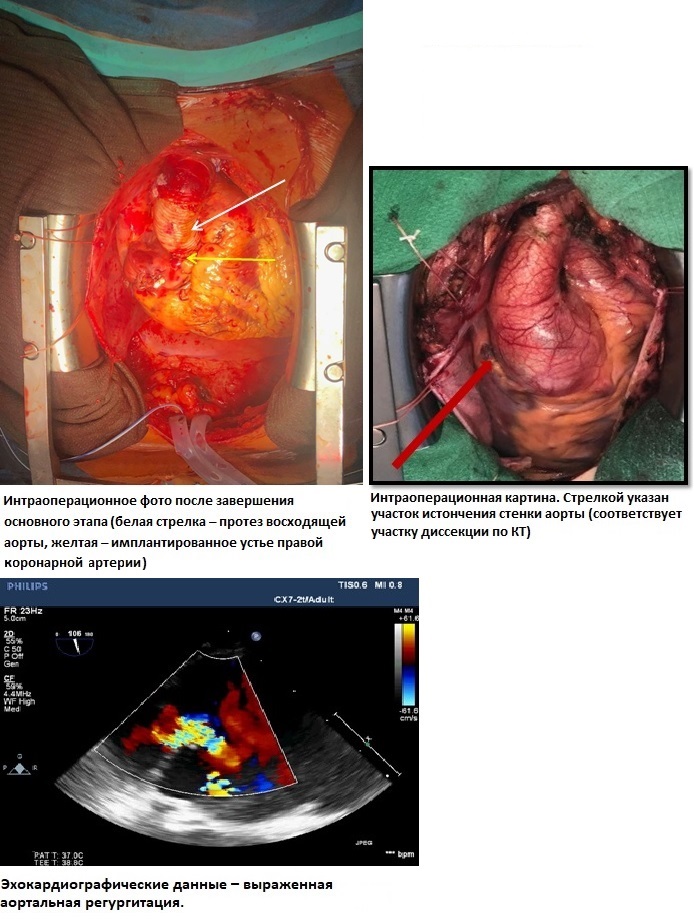
Искусственное кровообращение в условиях умеренной гипотермии до 29 градусов С в носоглотке. Дренирование левого желудочка через правую верхнюю легочную вену. Восходящая аорта пережата на 1 см проксимальнее устья брахиоцефального ствола под контролем церебральной оксиметрии. Кардиоплегия раствором «Кустодиол» антеградно селективно в устья коронарных артерий (1000 мл в левую коронарную артерию, 1000 мл в правую коронарную артерию). Подготовка аортального клапана к реимплантации: выделен корень аорты, вскрыт просвет аорты, выполнена ревизия аортального клапана. На комиссуры наложены швы–держалки, иссечена стенка аорты в зоне коронарных синусов, выделены устья коронарных артерий на «кнопках». Использован специально сформированный в виде синусов Вальсальвы синтетический протез BBraun Sinus 28 мм. Выполнена реимплантация аортального клапана в сосудистый протез: наложены швы на уровне фиброзного кольца изнутри наружу синтетической атравматической полиэстерной нитью 2–0 с прокладками с прошиванием проксимального края сосудистого протеза, произведена фиксация сосудистого протеза к фиброзному кольцу. Повторный пассаж кардиоплегии по 500 мл в устья коронарных артерий. Далее – моделирование высоты комиссур по отношению к сосудистому протезу для получения оптимальной коаптации аортальных створок, формирование гемостатических швов клапана, затем реимплантация устьев коронарных артерий на «кнопках» в модификации Kouchukos. Заключительный пассаж кардиоплегии. Между аортой и отдельным протезом 28 мм сформирован дистальный анастомоз по методике «погружения» внутрь аорты на 2 см проксимальнее брахиоцефального ствола. Проксимальный и дистальный протез сшиты между собой. Для дополнительной герметизации швом был использован хирургический клей BioGlue. Стандартное окончание ИК и операции (рис. 11).
Рис. 11 Схематичное изображение хода операции
Пациентка переведена в отделение реанимации на искусственной вентиляции легких, время искусственной вентиляции легких составило 15 часов, время пребывания в ОРИТ – 76 часов. Количество отделяемого по дренажам за первые сутки – 450 мл, за вторые сутки – 100 мл. На 4–е послеоперационные сутки пациентка в удовлетворительном состоянии переведена в профильное отделение.
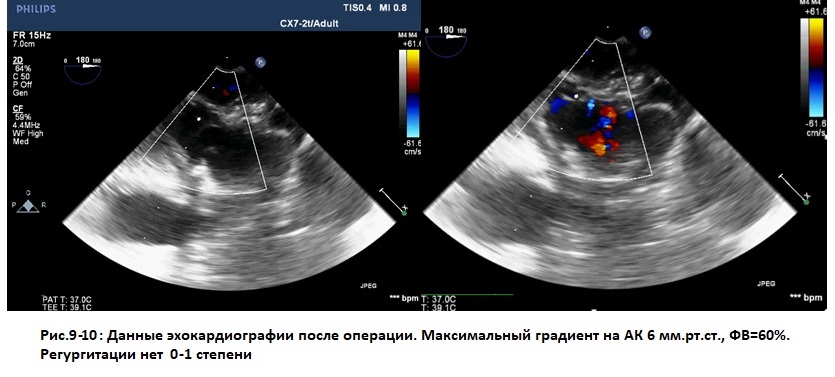
Послеоперационный период протекал без особенностей, послеоперационные раны заживали первичным натяжением, грудина стабильна. На 14-е послеоперационные сутки пациентка выписана под наблюдение кардиолога по месту жительства. По данным трансторакальной ЭХОКГ в послеоперационном периоде функция клапана удовлетворительная, максимальный градиент на аортальном клапане 6 мм рт.ст., регургитация до 1 ст. (рис. 9). По данным МСКТ с контрастированием, протез корня аорты на протяжении 48 мм, диаметр до 30 мм, восходящий отдел аорты до 21 мм, проксимальный отдел аорты до 21 мм (рис. 10).
Выводы
Современные технологии операций на корне аорты с сохранением нативного аортального клапана позволяют обеспечить благоприятные непосредственные функциональные результаты вмешательства и в перспективе могут улучшить прогноз и качество жизни у пациентов с синдромом Марфана и аневризмой восходящей аорты.
Литература
1. Медицинская генетика: учебник // В. Н. Запорожан, Ю. И., Бажора, А. В. Шевеленкова, М. М. Чеснокова. — Одесса: ОНМедУ, 2012. — 278 с. — (Серия «Библиотека студента–медика»). ISBN 978–966–443–004–0
2. Международные рекомендации диагностики синдрома Марфана — Гентские критерии (Ghent criteria, De Paepe A. et al., 1996, Loeys B. et al., 2010)
3. Всероссийское научное общество кардиологов. «Наследственные нарушения соединительной ткани», российские рекомендации, секция «Дисплазии соединительной ткани сердца», Москва, 2012
4. «Диагностика и лечение наследственных и многофакторных нарушений соединительной ткани». Национальные клинические рекомендации, Республика Беларусь, Минск, 2014 г.
5. Клиническая генетика: учебник // Г.Р.Мутовин – ГЭОТАР–Медиа, 2010 г.
6. David, T. E. (2016). Aortic Valve Sparing in Different Aortic Valve and Aortic Root Conditions. Journal of the American College of Cardiology, 68(6), 654–664.doi:10.1016/j.jacc.2016.04.062
7. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases; The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC)
8. Valve–sparing aortic root replacement in patients with Marfan syndrome-the Homburg experience// Ulrich Schneider, Tristan Ehrlich, Irem Karliova, Christian Giebels, Hans–Joachim Schäfers. Department of Thoracic and Cardiovascular Surgery, Saarland University Medical Center, Homburg/Saar, Germany
9. Long–term outcomes of aortic root operations for Marfan syndrome: A comparison of Bentall versus aortic valve–sparing procedures.// Joel Price, MD, et al – J Thorac Cardiovasc Surg. 2016 Feb;151(2):330–6. doi: 10.1016/j.jtcvs.2015.10.068. Epub 2015 Oct 27
10. Valve–Sparing Aortic Root Replacement: Early and Midterm Outcomes in 83 Patients. J.Coselli et al. The Annals of Thoracic Surgery, April 2014 Volume 97, Issue 4, Pages 1267–1274
Источник