No и пролапс митрального клапана синдром марфана

Причины синдрома Марфана. Пролапс митрального клапана
Синдром Marfan вызывается мутациями гена, кодирующего белок фибриллин-1 (FBN1), основной составной элемент микрофибрилл, которые являются компонентами внеклеточного матрикса, широко представленного в организме и выполняющего множество функций. Микрофибриллы и тропоэластин образуют эластические волокна.
Фрагментация и нарушение организации эластических волокон в медии аорты долгое время служили гистологическим маркером синдрома Marfan (неправильно называемого кистозной дегенерацией медии), хотя аналогичные микроскопические патологические повреждения встречаются при наследственной аневризме аорты и в аортах пожилых людей из нормальной популяции.
Дефекты в структуре миофибрилл приводят к аномальной секвестрации латентного TGF-бета-связывающего комплекса и повышению активности TGF-p; этим объясняют почти все плейотропные проявления синдрома Marfan.
В разных семьях описаны сотни различных мутаций гена FBN1, но лишь некоторые из них случайно проявляются у больных, не состоящих в родстве. В силу больших размеров гена FBN1 (9000 нуклеотидов в мРНК), расположенного в локусе хромосомы 15q21.1, содержащего 65 экзонов и занимающего участок ДНК > 240 тыс. п.н., поиск в нем мутации — непростая и недешевая задача.
Однако, как только мутация установлена, диагноз семейного заболевания становится очевидным. В семьях, члены которых поражены одним заболеванием, с целью клинической и пренаталыюй диагностики возможен анализ сцепления генов. Следует сказать, что наследуемая по аутосомно-домииантному типу эктопия хрусталика, характерный для членов семьи высокий рост, наличие фенотипа MASS (mitral valve, aorta, skin and skeletal abnormalities; митральный клапан, аорта, аномалии кожи и скелета) и наследственная аневризма аорты являются фенотипами, связанными с мутациями гена FBN1, и представляют собой именно те заболевания, которые интересуют клиницистов для исключения у пациентов неясного диагноза.
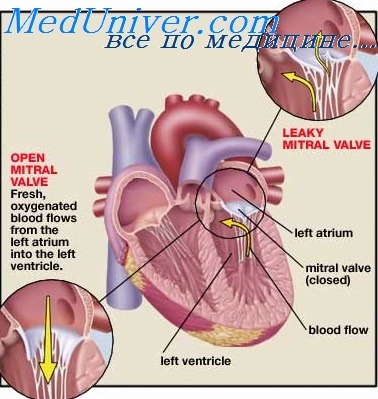
Пролапс митрального клапана
Это наиболее распространенное заболевание клапанов сердца человека, по причины и патогенез этого нарушения весьма гетерогенны. Совершенно очевидно, что пролапс митрального клапана не всегда является болезнью. Наследственные формы ПМК можно разделить на три группы. I группа — семейная форма с минимальным поражением экстракардиальных систем. II группа — клинически разнородное аутосомно-доминантное заболевание, крайней формой проявления которого являемся синдром Marfan; его можно рассматривать как дисплазию соединительной ткани. III группу составляют различные синдромы, которые включают 1IMK как вариант плейотропного проявления. Во всех этих группах ПМК часто сочетается с пролапсом трехстворчатого клапана.
К I группе наследственных форм пролапса митрального клапана, которые иногда называют синдромом пролапса митрального клапана, или семейным пролапсом митрального клапана, относят нарушения со стороны самого митрального клапана. Формирование реального ПМК сопровождается зависимыми от пола и возраста изменениями, характерными для идиопатической формы заболевания. Строгие генетические исследования с участием многих семей подтвердили аутосомно-доминантный характер наследования с переменной экспрессией.
Эта форма встречается у пациентов с деформирующимися во время тока крови створками митрального клапана и с избыточно расширяющимся во время систолы кольцом митрального клапана; поскольку такой фенотип воспроизводится довольно точно, могут существовать две различные аутосомно-доминантные формы заболевания. Причины этих состояний неизвестны. Более того, неясно, когда и как фенотипическое проявление такого состояния можно отличить от спорадических случаев ПМК и случаев доказанного системного заболевания соединительной ткани. Единственными четкими экстра-кардиальными признаками заболевания являются избыточный размах рук у женщин, а также относительно низкий вес и сниженное САД.
Пролапс митрального клапана картирован в трех генетических локусах; причем установлено, что один из генов кодирует филамин А (FMNA). Два других локализованы на аутосомах (неполовых хромосомах) 11 p15.4 и 16р11.2-р12.1. Подверженность всех клапанов сердца миксоматозной дегенерации может наследоваться как признак, сцепленный с Х-хромосомой, который картируется как Xq28; дефект затрагивает ген, кодирующий FMNA.
— Также рекомендуем «Пролапс митрального клапана при синдроме Marfan или Ehlers-Danlos. ПМК при синдроме истонченной дряблой кожи»
Оглавление темы «Генетические болезни сердечно-сосудистой системы»:
1. Причины синдрома Марфана. Пролапс митрального клапана
2. Пролапс митрального клапана при синдроме Marfan или Ehlers-Danlos. ПМК при синдроме истонченной дряблой кожи
3. Синдром Ehlers-Danlos. Эластическая псевдоксантома (ЭПК)
4. Поликистоз почек взрослых (ПКПВ). Артериопеченочная дисплазия — синдром Alagille
5. Наследственные аневризмы и расслоения артерий. Врожденная извитость артерий
6. Врожденное внутричерепное кровоизлияние. Наследственные окклюзии артерий
7. Наследственная гемиплегическая мигрень и легочная гипертензия. Наследственный атеросклероз
8. Наследственный варикоз и атрезия вен. Наследственная лимфедема
9. Врожденная геморрагическая телеангиэктазия (ВГТ). Болезнь Osler-Rendu-Weber
10. Синдром von Hippel-Lindau. Синдром удлиненного интервала QT
Источник

Синдромом, или болезнью Марфана называют наследственное аутосомно-доминантное заболевание соединительной ткани, сопровождающееся характерной триадой поражений опорно-двигательной, сердечно-сосудистой системы и органов зрения. В классических случаях лица с этой патологией имеют астенический тип телосложения, высокий рост, удлиненные «паучьи» пальцы и дефицит массы тела. Кроме этого, могут наблюдаться такие деформации костных структур как узкий лицевой скелет, плоскостопие, кифосколиоз, «готическое» нёбо и килевидная или воронкообразная грудная клетка.
Впервые данная наследственная патология была описана в 1876 году Вильямсом, а свое название заболевание получило 20 лет спустя по фамилии французского профессора и педиатра Антуана Марфана, который наблюдал девочку по имени Габриель с подобным симптомокомплексом. Этим синдромом страдали многие известные люди – скрипач Никколо Паганини, сказочник Ханс Христиан Андерсен, президент США Авраам Линкольн, композитор Сергей Рахманинов, певец Трой Сиван, первая подиумная модель Лесли Хорнби, многократный олимпийский чемпион Майкл Фелпс и многие другие. Такая частая встречаемость данного заболевания среди знаменитостей даже вызвала у ученых предположение, что одним из компонентов генетических отклонений являются гениальные способности.
Однако почти все проявления синдрома Марфана приводят к тяжелым инвалидизирующим последствиям, и одним из кардинальных признаков этого наследственного недуга являются патологии сердца и сосудов. Примерно в 90-95% случаев именно они становятся причиной смерти больных в возрасте 40-50 лет. Развитие современной кардиохирургии позволяет во многих случаях увеличивать продолжительность жизни таких пациентов до 60-70 лет. Кроме этого, вовремя выполненная коррекция сердечно-сосудистых патологий существенно улучшает качество жизни больных.
В этой статье мы ознакомим вас с проявлениями синдрома Марфана со стороны сердца и сосудов и способами диагностики и лечения этих опасных для жизни патологий. Эта информация поможет вам понять суть этого недуга, и вы сможете задать возникающие у вас вопросы своему лечащему врачу.
Симптомы
 Синдром Марфана часто сопровождается аневризмой аорты
Синдром Марфана часто сопровождается аневризмой аорты
Со стороны сердца и сосудов при болезни Марфана наблюдаются следующие патологии:
- дилатация и аневризмы аорты;
- клапанные пролапсы (особенно часто пролапс митрального клапана);
- «висячее», или «капельное» сердце.
По разным данным статистики, патологии аорты при этом синдроме наблюдаются в 65-100% случаев, а пролапс митрального клапана – у 61-100% пациентов. Кроме этого, у больных часто выявляется дисфункция левого желудочка.
При неонатальной форме синдрома признаки недуга появляются сразу после рождения ребенка и вызывают быстрое прогрессирование сердечной недостаточности, приводящей к наступлению смерти до 1 года жизни. У детей с такой патологией во внутриутробном периоде часто формируются врожденные пороки сердца и сосудов – дефекты межжелудочковой или межпредсердной перегородки, стеноз легочной артерии и коарктация аорты. Впоследствии сердечно-сосудистые патологии нередко вызывают нарушения ритма и развитие инфекционного перикардита.
Патологии аорты
При синдроме Марфана наиболее часто происходит дилатация корня аорты. Обычно его значительное расширение происходит к 13 годам. Дилатация развивается постепенно, симметрично и наиболее выражена среди мальчиков. Изолированные расширения часто протекают бессимптомно, но в некоторых случаях сопровождаются болями в груди, возникающими во время физической активности.
Иногда дилатация происходит на уровне восходящей аорты или синусов Вальсальвы. При расширении аорты и значительном истончении ее стенки на уровне синусов у больного возникают аневризмы синусов Вальсальвы. Чаще поражается коронарный правый синус, реже – левый коронарный или некоронарный синус. До прорыва такие аневризмы никак себя не проявляют и обычно выявляются случайно во время Эхо-КГ. Иногда такие сосудистые выпячивания проявляются болями и аритмиями, а при сильном выбухании аневризмы происходит обструкция выходного отдела правого желудочка.
Прорывы аневризм могут вызываться следующими факторами:
- физическая активность;
- повышение артериального давления;
- развитие бактериального эндокардита.
При разрыве таких аневризм внезапно появляются следующие симптомы:
- боль в сердце;
- учащенный пульс;
- одышка.
При осмотре больного и выслушивании тонов сердца определяются:
- «машинный» систолодиастолический шум над сердцем;
- диастолическая гипотензия;
- систолическое дрожание.
В более редких случаях аневризмы аорты возникают в таких ее отделах как дуга, восходящая или нисходящая часть, брюшной отдел. При формировании аневризмы в грудном отделе аорты у больного могут возникать сдавления трахеи, вызывающие острые дыхательные нарушения. В некоторых редких случаях при синдроме Марфана формируются множественные аневризмы. Иногда они поражают левую коронарную артерию.
Расслоение аорты может вызывать ее внезапный разрыв или протекает хронически, приводя к формированию гигантской аневризмы. Клинические проявления аневризм будут зависеть от места расположения таких выпячиваний и степени компрессии окружающих их тканей и органов:
- на восходящей аорте – отечность лица, головные боли;
- на дуге аорты – затруднения при глотании, осипший голос, рефлекторно возникающий кашель, боли за грудиной и между лопатками;
- на торакоабдоминальных отделах – боли, ощущения дискомфорта, тяжести, пульсации в животе.
Разрывы аневризм провоцируют развитие шока и сердечно-сосудистого коллапса. Кроме этого, у больного возникают симптомы, кровотечения в тот или иной орган – легкие, плевральную полость, забрюшинное пространство.
Аортальная недостаточность, сопровождающаяся несостоятельностью аортального клапана и забросом крови в левый желудочек, у больных с синдромом Марфана вызывается дилатацией аорты, аннулоаортальной эктазией и поражением митрального клапана, характеризующимся миксоматозной дегенерацией его створок, кальцинированием клапанного кольца и патологическим удлинением и разрывом хорд створок. В результате у больного происходит аортальная регургитация, объем которой тесно связан с размером корня аорты:
- при диаметре корня 4 см – происходит минимальная регургитация;
- при диаметре корня 5 см – происходит регургитация I-II степени у 81% больных;
- при диаметре корня 6 см и более – происходит регургитация II степени у всех больных.
Иногда возникающая при синдроме Марфана аортальная регургитация, вызывается инфекционным поражением аортального клапана.
При острой регургитации у больного развивается кардиогенный шок, проявляющийся следующими симптомами:
- выраженная слабость;
- резкое снижение артериального давления;
- одышка;
- отек легкого.
При хронической аортальной регургитации у больного сначала возникает одышка. Впоследствии появляются признаки стенокардии напряжения, которые могут проявляться и в ночное время. Больной ощущает боли в груди, страх смерти, у него появляется холодный липкий пот. Пациенты часто жалуются, что им тяжело переносить сердцебиение и боль в груди в положении лежа. Такое проявление аортальной недостаточности без проведения кардиохирургической коррекции может приводить к быстрому прогрессированию сердечной недостаточности и наступлению смерти.
Пролапс митрального клапана
Пролапс митрального клапана также может быть симптомом синдрома Марфана
При синдроме Марфана часто наблюдается пролапс митрального клапана, сопровождающийся выбуханием клапанных створок (одной или обеих) в полость левого предсердия. Такой порок сердца проявляется возникновением характерных шумов в сердце:
- изолированные среднесистолические щелчки;
- сочетание позднего систолического шума со щелчками;
- голосистолический шум.
Впоследствии митральный пролапс начинает сопровождаться регургитацией и может осложняться тромбоэмболиями и инфекционным эндокардитом. Острая форма митральной регургитации провоцируется отрывом сухожильных нитей от клапанной створки, а хроническая – неполноценным смыканием клапанных створок и значительным расширением атриовентрикулярного фиброзного кольца.
Впоследствии любая форма митральной регургитации приводит к дисфункции левого желудочка, застойной сердечной недостаточности и предсердным аритмиям. Особенно тяжело данная патология сердца протекает именно в детском возрасте, так как у детей ей часто сопутствует функциональная атрезия трехстворчатого клапана.
Дисфункция левого желудочка
Еще одним проявлением поражений сердца при болезни Марфана становится дисфункция левого желудочка, которая проявляется в форме дилатационной кардиомиопатии и сокращении сократительной способности сердца. Она не всегда связана с присутствием пороков клапанного аппарата и может наблюдаться и при отсутствии митральной или аортальной регургитации.
Из-за левожелудочковой дисфункции возникают следующие симптомы:
- быстрая утомляемость;
- одышка;
- затруднение дыхания в положении лежа;
- приступы стенокардии;
- эпизоды сердечной астмы.
При развитии застоя в большом круге кровообращения у больного появляются ощущения тяжести в правом подреберье, отеки ног и скопление жидкости в брюшной полости (асцит).
Если дисфункция левого желудочка сопровождается аритмиями, то у больного возникают ощущения перебоев в работе сердца, эпизоды головокружений и обморочных состояний. Иногда дилатационная кардиомиопатия приводит к развитию таких осложнений как тромбоэмболии и инсульт, которые могут становиться причиной внезапной смерти.
На ЭКГ у больных с синдромом Марфана выявляются признаки:
- гипертрофии левого желудочка;
- различных аритмий;
- нарушений процесса реполяризации (синдром WPW);
- внутрижелудочковые и AV-блокады.
В последние годы специалисты предполагают, что при синдроме Марфана присутствует и нарушение нейровегетативной регуляции тонуса сосудов и деятельности сердца. Многими авторами описаны случаи ортостатической гипотензии, которая часто проявляется головными болями. Эти симптомы вызываются диффузным расширением периферических сосудов, и эти данные подтверждаются при выполнении МРТ.
Диагностика
Характер и степень тяжести патологий сосудов и сердца при синдроме Марфана оценивается по данным следующих обследований:
- ЭКГ (обычное и по Холтеру);
- Эхо-КГ;
- рентгенография грудной клетки;
- МРТ и КТ сердца.
При подозрении на аневризму и расслоение аорты показано проведение аортографии.
По результатам всех данных инструментальных обследований составляется дальнейший план лечения пациента и принимается решение о необходимости выполнения кардиохирургической коррекции.
Лечение
 Характер нарушений сердечно-сосудистой системы при синдроме Марфана определяют при помощи дополнительных методов исследования, в частности Холтеровского мониторирования
Характер нарушений сердечно-сосудистой системы при синдроме Марфана определяют при помощи дополнительных методов исследования, в частности Холтеровского мониторирования
Детям с синдромом Марфана рекомендуется строго дозированная физическая нагрузка с исключением занятий контактными и силовыми видами спорта. Они должны заниматься физкультурой в специализированных группах. Кроме этого, запрещается ношение тяжестей, сельскохозяйственный труд, перегревания и походы на длительные дистанции. В дальнейшем им не следует выбирать специальности, связанные с работами на вредных производствах (химические испарения, радиация, высокие температуры) и профессии, требующие значительных физических или психоэмоциональных затрат, высокой остроты зрения или связанные с вибрацией.
При планировании зачатия ребенка женщина должна знать о том, что ей будет необходимо наблюдаться у сосудистого хирурга и каждые 2 месяца проводить Эхо-КГ, а при расширении аорты более 45 мм может возникать вопрос о целесообразности дальнейшего сохранения беременности. Кроме этого, усугубляющееся расширение аорты может становиться показанием для выполнения кардиохирургической коррекции. Для родоразрешения таким женщинам рекомендуется кесарево сечение, выполняющееся в специализированных стационарах для рожениц с патологией сердца и сосудов.
В качестве адекватной физической нагрузки, исключать которую полностью нельзя для сохранения физического и психологического комфорта, лицам с болезнью Марфана показаны нагрузки и спорт, не связанные с резкими и активными усилиями. Это может быть езда на велосипеде, запуск радиоуправляемых моделей, ходьба, рыбалка и т. п.
При синдроме Марфана рекомендуется включать в свой рацион большее количество продуктов, богатых магнием, или принимать препараты на его основе. По мнению многих специалистов, диета с высоким содержанием магния способствует более медленному развитию патологических изменений сердца и сосудов.
Медикаментозная терапия
При синдроме Марфана для предупреждения дальнейшей дилатации аорты назначаются b-адреноблокаторы, способствующие уменьшению нагрузки на стенки аорты и корректирующие артериальную гипертензию. Для этого могут использоваться Пропранолол или длительнодействующие b-адреноблокаторы (Атенолол и пр.). В последние годы была доказана эффективность назначения ингибиторов АПФ (Эналаприла и др.) и антагонистов кальция (Нифедипина, Верапамила и др.), которые так же способны предупреждать дальнейшее расширение аорты.
При необходимости проведения хирургических манипуляций и санации зубов людям с болезнью Марфана рекомендуется в профилактических целях принимать антибиотики, предупреждающие развитие инфекционного эндокардита.
Хирургическое лечение
Показаниями для хирургического лечения при расслоении или аневризмах аорты становятся следующие клинические случаи:
- хроническое расслоение аортальной стенки;
- расширение корня аорты до более 55 мм и относительная аортальная регургитация;
- аортальная регургитация, сопровождающаяся выраженной левожелудочковой дисфункцией.
План кардиохирургической коррекции составляется на основании данных селективной аортографии. После этого выполняются реконструктивные операции на аорте, заключающиеся в замене расширенной части сосуда эндо- или экзопротезами. При протезировании аневризмы корня аорты используются клапан-содержащие протезы, а при присутствии аннулоаортальной эктазии одновременно выполняется протезирование дуги и корня аорты. В некоторых случаях кардиохирургические операции могут дополняться заменой митрального клапана.
Ранее такие вмешательства часто приводили к осложнениям или послеоперационной смертности. Однако внедрение в кардиохирургию новых методик позволило сократить процент подобных нежелательных последствий, и после удачного протезирования больные становятся вполне трудоспособными.
Проявления синдрома Марфана со стороны сердца и сосудов нуждаются в постоянном наблюдении сосудистых хирургов. Больные с этим заболеванием должны соблюдать все рекомендации врача и регулярно проходить обследования для отслеживания динамики патологии. Для коррекции возникающих нарушений могут применяться консервативные методы терапии или хирургическое лечение. Кроме этого, больные с данной патологией должны постоянно наблюдаться у офтальмолога и ортопеда.
Первый канал, программа «Жить здорово!» с Еленой Малышевой, в рубрике «Про медицину» разговор о синдроме Марфана (см. с 30:20):
Источник