Мукополисахаридоз 1 типа код по мкб 10

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Содержание
- Описание
- Симптомы
- Лечение
- Прогноз
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Мукополисахаридоз.
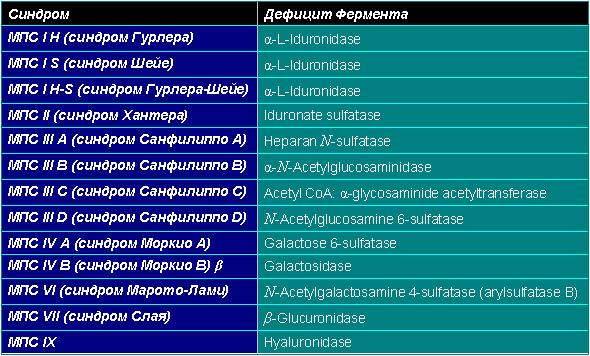
Дефицит ферментов при мукополисахаридозах
Описание
Мукополисахаридозы – это группа наследственных заболеваний, вызванных неполным разрушением и накоплением кислых мукополисахаридов (гликозоаминогликанов).
Клиника обусловлена накоплением их в различных органах. Мукополисахариды играют важную роль в поддержании упругости и целостности соединительной ткани, упругости хряща. Мукополисахаридозы наследуются по аутосомно-рецессивному типу, за исключением синдрома Гунтера. Существует несколько типов заболевания, различающихся по биохимическому дефекту.
Симптомы
Мукополисахаридоз 1 типа – синдром Гурлера. Характеризуется дефицитом фермента альфа-идуронизады, что приводит к накоплению продуктов метаболизма в тканях и их эскреции с мочой. Во всех тканях расположены вакуолизированные клетки, содержащие лизосомы, переполненные мукополисахаридами. В клетках головного мозга накапливаются липиды.
Дети с синдромом Гурлера в момент рождения выглядят здоровыми и клинические проявления появляются на втором году жизни. При объективном обследовании выявляют гепатоспленомегалию, усиленный кифоз, постоянные выделения из носа, шумное дыхание. Появляются дисморфические изменения: выпуклый и нависающий лоб, плоский нос с запавшей переносицей, грубые и утолщенные губы, гипертелоризм. Волосы на голове жесткие и густые. Язык увеличен, зубы мелкие, ушные раковины деформированы. Голос хриплый. Туловище короткое, грудная клетка деформирована, выражен грудной и пояснично-грудной кифоз. Эпифизы длинных костей утолщены и расширены, окостенение нарушено. Имеются проявления порока сердца. Живот увеличен, имеют место гепатомегалия и пупочная грыжа. Пальцы кистей находятся в полусогнутом положении, ограничена подвижность суставов. У большинства больных наблюдается помутнение роговицы, катаракта, врожденная глаукома. Интеллект страдает, больные страдает, больные отстают в умственном развитии.
Мукополисахаридоз 2 типа – синдром Гунтера. Это единственный мукополисахаридоз, сцепленный с Х-хромосомой. Встречается у мальчиков. Течение его более доброкачественное. Развитие этого заболевания связано с недостаточностью фермента идуронатсульфатазы. Существует тип А и тип Б этого заболевания.
Тип А – классическая форма синдрома Гунтера. Для нее характерны грубые черты лица, низкорослость, тугоподвижность суставов, гепатоспленомегалия, грыжи, выраженная умственная отсталость. Заболевание прогрессирует медленнее. Часто у больных утрачен слух. Нередки кожные проявления, заключающиеся в появлении папул на коже спины. В патологический процесс часто вовлекается сердце. Может отмечаться умеренный кифоз. Продолжительность жизни – около 20 лет.
Тип Б – значительно более доброкачественный, продолжительность жизни у больных большая.
Мукополисахаридоз 3 типа – синдром Санфилиппо. Сопровождается эскрецией с мочой гепаринсульфата, который также накапливается в тканях. Заболевание проявляется самой глубокой прогрессирующей задержкой умственного развития. Клинические проявления ярко выражены у детей раннего возраста. Ребенок отстает в развитии, гиперактивен. До 10 лет идет быстрое прогрессирующее ухудшение состояния. Большинство детей умирает в середине второго десятилетия жизни. К характерным симптомам заболевания относятся умственная отсталость, тугоподвижность суставов, гепатоспленомегалия, грыжи, множественный дизостоз.
Мукополисахаридоз 4 типа – синдром Моркио. Обусловлен дефицитом ферментов гаактозо-6-сульфатазы и галактозамин-6-сульфатазы. На 2-ом году жизни дети начинают отставать в росте и у них появляется скелетные деформации (вальгусная деформация коленных суставов, выбухание нижних ребер, кифосколиоз). Степень укорочения туловища превышает укорочение конечностей. Интеллект относительно сохранен. Отмечается задержка физического развития, помутнение роговицы, выступающая нижняя часть лица, гипоплазия эмали зубов, короткая шея, килевидная грудная клетка, поясничный лордоз. У таких детей большой живот, гиперподвижность и подвывихи суставов, плоскостопие, отмечается снижение слуха. К 20 годам выявляется регургитация аорты. Смерть к этому возрасту обычно наступаетот сердечной и неврологической симптоматики (вследствие сдавления спинного мозга деформированными позвонками). Диагностика основана на выявлении в моче кератансульфата или кислых мукополисахаридов. Тип наследования аутосомно-рецессивный.
Агрессивность.
Внешний вид больной с мукополисахаридозом
Лечение
До сих пор не существует эффективного лечения синдрома Гурлера, в основном применяется симптоматическое и ортопедическое терапия. Больные обычно умирают в подростком возрасте.
Лечение заболевания заключается в назначении трансфузий крови, плазмы и лейкоцитарной массы. Показано применение глюкокортикоидов, тиреоидина, витамина А, витаминов группы В.
Прогноз
Прогноз при всех типах мукополисахаридозов неблагоприятный, болезнь неуклонно прогрессирует, и летальный исход может наступить в любом возрасте. Нередки случаи смерти больных от сопуствующей патологии сердца и присоединения инфекционного агента.
Профилактика
Так как в настоящее время нет эффективной терапии мукополисахаридозов, то необходимо выявлять заболевание в неонатальном периоде. Целесообразным представляется проведение трансабдоминального амниоцентеза у беременных женщин с высоким уровнем риска рождения детей с мукополисахаридозом. Культивирование клеток амниотической жидкости и определение в них активности ферментов, расщепляющих гликозамингликаны, позволяет проводить адекватную антенатальную диагностику мукополисахаридозов в ранние сроки беременности (9-12 недель). Не менее важным компонентом профилактики мукополисахаридозов представляется проведение селективного скрининга с целью обследования некоторых контингентов больных: людей с низким зрением, с задержкой умственного развития, нарушениями опорно-двигательного аппарата.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: E76.0
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E70-E90 Нарушения обмена веществ / E76 Нарушения обмена глюкозаминогликанов
Определение и общие сведения[править]
Мукополисахаридоз IH типа
Синонимы: Синдром Гурлер
Синдром Гурлер является наиболее тяжелой формой мукополисахаридоза 1 типа — это редкий вариант лизосомных болезней накопления. Характерезуется скелетными аномалиями, когнитивными нарушениями, заболеваниями сердца и дыхательных путей, увеличением печени и селезенки, характерным лицом и уменьшением продолжительности жизни.
Распространенность в Европе оценивается в 1 / 200,000. Мукополисахаридоз IH типа — составляет 57% всех случаев мукополисахаридоза 1 типа. Наследуется аутосомно-рецессивно.
Синдром Гурлер описала в 1919 г. педиатр Гертруда Гурлер (Германия).
Этиология и патогенез[править]
Вызывается мутациями гена IDUA (4p16.3), что приводит к полному дефициту фермента лизосом альфа-L-индуронидазы и накоплению дерматансульфата и гепарансульфата.
Клинические проявления[править]
Мукополисахаридоз IH типа (синдром Гурлер, синдром Гурлер-Пфаундлера)
проявляется на первом году жизни увеличением и деформацией черепа (макроцефалия, скафоцефалия — ладьевидный череп), искривлением позвоночника,
расширением венозной сети на висках, задержкой физического и умственного развития. Со временем формируются характерные признаки: запавшая переносица, гипертелоризм, помутнение роговицы, пигментная ретинопатия, увеличение языка и губ, кроткая шея, воронкообразная или килевидная грудь, паховые и пупочные грыжи, ограничение подвижности суставов. Рост замедляется со второго года жизни. Часты воспалительные заболевания верхних дыхательных путей, пневмония. В неврологическом статусе: гипертензионно-гидроцефальный синдром, олигофрения, двигательные и вегетативно-трофические расстройства. На краниограммах изменения величины и формы черепа, расхождение черепных швов, уплотнение костей черепа, уплощение и расширение турецкого седла. На ЭЭГ — угнетение альфа-ритма, единичные дельта- и тета-волны, острые волны, спайкоподобные колебания, дизритмия, неустойчивость амплитуды колебаний потенциалов. При поясничном проколе ликворное давление свыше 300 мл вод. ст., состав ликвора нормален, возможно лишь некоторое снижение содержания белка. К 2-3 годам дети произносят только отдельные слова, но и эти навыки могут быть нестойкими, так как прогрессирует слабоумие, нарастают и неврологические расстройства: изменения мышечного тонуса, повышение сухожильных рефлексов, пирамидные знаки, легкие нарушения координации. Возможны слепота, глухота. Обычны бледная, сухая, грубая кожа, пороки сердца, гепатоспленомегалия, клювовидная форма тел позвонков
Мукополисахаридоз, тип I: Диагностика[править]
Повышена экскреция с мочой дерматансульфата и гепарансульфата. В части лейкоцитов (до 60%) метахроматические гранулы. Дородовая диагностика возможна.
Дифференциальный диагноз[править]
Дифференциальный диагноз включают мягкую форму мукополисахаридоза 1 типа — синдром Гурлер-Шейе, мукополисахаридоз 2, мукополисахаридоз 6 и муколипидоз 2.
Мукополисахаридоз, тип I: Лечение[править]
Трансплантация гемопоэтических стволовых клеток (ГСК) является методом выбора при лечении больных с синдромом Гурлер в возрасте менее 2,5 лет, а у отдельных пациентов и старше, она продлевает выживание, сохраняет когнитивные функции и улучшает некоторые соматические функции. Пожизненная ферментозаместительная терапия ларонидазой рекомендуется для всех пациентов, она купирует неневрологические симптомы.
Дополнительное поддерживающее лечение включает в себя хирургические вмешательства (аденотозиллэктомия, удаление грыжи, вентикулоперитонельный шунт, замена сердечного клапана, кистевой туннельный выпуск, спинальная декомпрессия), физическая, профессиональные и речевая терапия, респираторная поддержка, слуховые аппараты, анальгетики и препараты от желудочно-кишечных расстройств.
Прогноз
Пациенты часто умирают в первое десятилетие жизни от респираторных и сердечно-сосудистых осложнений, но современная терапия может значительно улучшить продолжительность жизни, для нее выжны ранние постановки диагноза и начало терапии.
Профилактика[править]
Прочее[править]
Синдром Гурлер-Шейе
Синонимы: Мукополисахаридоз 1H/S
Определение и общие сведения
Синдром Гурлер-Шейе это промежуточная форма мукополисахаридоза 1 типа, занимает положение между этим синдромом Гурлер и синдромом Шейе. Мукополисахаридоз 1H/S — это редкий вариант лизосомных болезней накопления, характеризуется деформациями скелета и задержками в развитии моторики.
На долю синдрома Гурлер-Шейе приходится 23% случаев мукополисахаридоза 1 типа с распространенностью приблизительно 1 / 435,000. Наследуется аутосомно-рецессивно.
Этиология и патогенез
Вызывается мутациями гена IDUA (4p16.3), что приводит к частичному дефициту фермента лизосом альфа-L-индуронидазы и накоплению дерматансульфата и гепарансульфата.
Клинические проявления
У больных с синдромом Гурлер-Шейе нормальный или почти нормальный интеллект, но проявляют различные степени физического ущерба. Пациенты демонстрируют низкорослость, множественные дизостозы, торакальный поясничный кифоз, прогрессирующее укрупнение черт лица разной степени тяжести, кардиомиопатию и клапанную патологию, нейросенсорную потерю слуха, увеличенные миндалины и аденоиды и назальную секрецию. Гидроцефалия может развиваться в возрасте двух лет. Помутнение роговицы наблюдается в возрасте от двух до четырех лет и требует проведения кератопластики. Другие симптомы могут включать в себя органомегалию, грыжи и гирсутизм.
Диагностика
Ранняя диагностика затруднена, поскольку первые клинические признаки не являются специфичными. Диагноз основан на обнаружении повышенной мочевой секреции гепарана и дерматансульфата и демонстрации ферментативного дефицита в культуре лейкоцитов или фибробластов. Генетическое тестирование доступно. Дородовая диагностика возможна.
Дифференциальный диагноз
Дифференциальный диагноз включают другие формы мукополисахаридоза 1 типа — синдромы Гурлер и Шейе, мукополисахаридоз 2, мукополисахаридоз 6 муколипидоз 2.
Лечение
Трансплантация гемопоэтических стволовых клеток (ГСК) является методом выбора при лечении больных с синдромом Гурлер в возрасте менее 2,5 лет, а у отдельных пациентов и старше, она продлевает выживание, сохраняет когнитивные функции и улучшает некоторые соматические функции. Пожизненная ферментозаместительная терапия ларонидазой рекомендуется для всех пациентов, она купирует неневрологические симптомы.
Прогноз
Ожидаемая продолжительность жизни при синдроме Гурлер-Шейе может быть сокращена, смерть наступает до подросткового возраста из-за серьезных сердечно-сосудистых и респираторных осложнений.
Синдром Шейе
Синонимы: Мукополисахаридоз 1S
Определение и общие сведения
Синдром Шейе это мягкая форма мукополисахаридоза 1 типа, относится к лизосомным болезням накопления, характеризуется деформациями скелета и задержками в развитии моторики.
Распространенность оценивается в 1/500000. На долю синдрома Шейе приходится 20% случаев мукополисахаридоза 1 типа. Наследуется аутосомно-рецессивно.
Этиология и патогенез
Вызывается мутациями гена IDUA (4p16.3), что приводит к частичному дефициту фермента лизосом альфа-L-индуронидазы и накоплению дерматансульфата и гепарансульфата.
Клинические проявления
Симптомы обычно возникают в возрасте до 5 лет, но настолько мягкие, что диагноз часто не рассматривается вплоть до взрослой жизни. Пациенты почти нормальной роста и не имеют интеллектуального дефицита. Помутнение роговицы происходит поступательно и диффузно, как правило, в возрасте четырех лет. Глаукома наблюдается чаще, чем при синдроме Гурлер. Пациенты демонстрируют мягкое огрублением черт лица — большой рот с толстыми губами. Пациенты могут страдать от назальной секреции, нейросенсорной потери слуха, ригидности суставов, мягких скелетных изменений и кистевого туннельного синдромома. Может присутствовать поражение аортального клапана. Компрессия шейного отдела спинного мозга, вызванная инфильтрацией гликозаминогликанами твердой мозговой оболочки, что может приводить к спастическим параличам.
Диагностика
Ранняя диагностика затруднена, поскольку первые клинические признаки не являются специфичными. Диагноз основан на обнаружении повышенной мочевой секреции гепарана и дерматансульфата и демонстрации ферментативного дефицита в культуре лейкоцитов или фибробластов. Генетическое тестирование доступно. Дородовая диагностика возможна.
Дифференциальный диагноз
Дифференциальный диагноз включают более тяжелые формы мукополисахаридоза 1 типа — синдромы Гурлер и Гурлер-Шейе, мукополисахаридоз 2, мукополисахаридоз 6 и муколипидоз 2.
Лечение
Ферментозаместительная терапия ларонидазой рекомендуется для всех пациентов, ее следует начинать при постановке диагноза и она может быть полезной у пациентов, ожидающих трансплантации гемопоэтических стволовых клеток (ГСК). Раннее лечение замедляет прогрессирование заболевания.
Прогноз
Средняя продолжительность жизни пациентов с синдром Шейе может быть слегка снижена.
Источники (ссылки)[править]
Офтальмоневрология [Электронный ресурс] / А. С. Никифоров, М. Р. Гусева — М. : ГЭОТАР-Медиа, 2014. — https://www.rosmedlib.ru/book/ISBN9785970428177.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Ларонидаза
Источник
Мукополисахаридо́з I (мукополисахаридоз типа I, МПС-I, англ. mucopolysaccharidosis I, MPS-I) — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов (англ. GAG или мукополисахаридов), вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Классификация[править | править код]
Согласно Международной классификации болезней десятого пересмотра (МКБ-10), различают:
- E7676. Нарушения обмена гликозаминогликанов:
- E76.076.0 Мукополисахаридоз, тип I. Синдромы: «Гурлер» (MPS-I H), «Шейе» (MPS-I S), «Гурлер — Шейе» (MPS-I H/S).
Встречающийся в литературе термин «гаргоилизм», введенный в клинику английским врачом Эллисом (англ. R. W.В. Ellis) в 1936 году до открытия биохимической основы патологического процесса[1], объединяет мукополисахаридозы типа I (Н, S, H/S) и типа II (синдром Хантера).
Патогенез[править | править код]
Недостаточность альфа-L-идуронидазы (фермента лизосом) способствует накаплению мукополисахаридов одного из двух классов: гепарансульфата или дерматансульфата[1].
Клиническая картина[править | править код]
Несмотря на то, что к развитию муколисахаридоза I типа ведёт дефицит одного фермента альфа-L-идуронидазы, заболевание проявляется в трёх клинических вариантах: синдром Гурлер, синдром Шейе и синдром Гурлер — Шейе.
Исторически первым двумя педиатрами: австрийским — нем. Hurler Gertrud [G. Hurler] (1889–1965) и немецким — нем. Pfaundler Meinhard von [M. V. Pfaundler] (1872–1947)[2] описан синдром, при котором определяется подавляющее большинство компонентов фенотипа, характерных для этой группы наследственных заболеваний, и все они — резко выражены. Описанная авторами болезнь, ставшая прототипом всех мукополисахаридозов I типа, проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Позже американским офтальмологом Шейе, англ. Н. G. Scheie (1909–1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе[2]. Последней описана промежуточная форма болезни, названная синдром Гурлер — Шейе[3].
Синдром Гурлер[править | править код]
Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается с конца первого года жизни). Характерны признаки гаргоилизма: крупный череп, крутой лоб, запавшая переносица, толстые губы, большой язык, характерное выражение лица («лицо выплевывающего воду»). Кроме того, отмечается короткая шея, ограниченная подвижность суставов (тугоподвижность, главным образом, затрагивает локтевые и межфаланговые суставы пальцев кистей и стоп), фиксированный кифоз на месте перехода грудных позвонков к поясничным, укорочение конечностей, в основном, происходит за счёт проксимальных отделов (бёдер и плеч), в меньшей степени — голеней и предплечий. Весьма своеобразно строение кисти пациента: пальцы короткие, одинаковые по длине (изодактилия) расходятся веерообразно, напоминая трезубец. Нижнепоясничный лордоз способствует выпячиванию живота вперёд, а ягодиц назад. Отмечается гепатоспленомегалия, склонность к формированию пупочной грыжи. Характерно диффузное помутнение роговицы за счёт накопления в ней дерматансульфата. Возможно развитие слабоумия, кариеса зубов, характерной формы ногтевых пластинок в виде часовых стёкол, тугоухость или глухота, низкий хриплый голос, гипертрихоз, сухие и жёсткие волосы. В большинстве случаев в патологический процесс вовлекается сердце — оно увеличивается в размерах, происходят изменения клапанов, миокарда, эндокарда, крупных и коронарных артерий. При рентгенологическом исследовании определяется преждевременное окостенение лямбдовидного шва, расширение турецкого седла, характерная форма позвонков («рыбьи позвонки»), искривление лучевой кости, деформации метафизарных и эпифизарных отделов длинных трубчатых костей, короткие метакарпальные кости и фаланги пальцев. Такие дети обычно не доживают до 10 лет[4].
Синдром Шейе[править | править код]
Клиническая симптоматика развивается на фоне нормальной продолжительности жизни[5][6], обычно не проявляется до достижения возраста 4 — 5 лет и может включать в себя[7]:
- помутнение роговицы с прогрессирующей потерей зрения вплоть до слепоты;
- гаргоилизм: грубые черты лица, широкий рот с полными губами, прогнатизм;
- избыточный рост волос на теле (гипертрихоз, гирсутизм);
- характерные деформации кистей и стоп;
- тугоподвижность суставов.
Синдром Гурлер — Шейе[править | править код]
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее тяжёлым промежуточным вариантом между синдромами Гурлер и Шейе. Фенотип пациентов также является промежуточным между фенотипом свойственным синдрому Гурлер и Шейе. Существует предположение, что больные синдромом Гурлер — Шейе являются генетическими химерами с одним аллелем синдрома Гурлер и вторым — синдрома Шейе[1].
Клиническая симптоматика характеризуется в основном кожными проявлениями, сочетающимися с умеренной умственной отсталостью и помутнением роговицы[3].
Диагностика[править | править код]
- Определение активности лизосомальных гидролаз (альфа-L-идуронидазы);
- Исследование мочи — определение экскретируемого с мочой типа мукополисахарида (дерматансульфат, гепарансульфат).
Лечение[править | править код]
Современная наука получила возможность проводить фермент-заместительную терапию для пациентов с мукополисахаридозом I типа, вызываемого дефектом альфа-L-идуронидазы (англ. α-L-iduronidase, lauonidase) — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов[8], составляющих основу межклеточного вещества соединительной ткани. Группа мукополисахаридоза I типа включает в себя пациентов с синдромом Гурлер (мукополисахаридоз-I H), синдромом Шейе (мукополисахаридоз-I S), а также синдромом Гурлер — Шейе (мукополисахаридоз-I H/S). Ранняя диагностика и своевременная терапия развившейся компрессии спинного мозга может предотвратить необратимые повреждения нервов. Лечение также назначается пациентам с проблемами внутрисердечной гемодинамики, вызванными недостаточностью клапанов[7].
См. также[править | править код]
- Мукополисахаридоз
- Генные болезни
- Лизосомные болезни накопления
Примечания[править | править код]
- ↑ 1 2 3 Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения 13 декабря 2014.
- ↑ 1 2 Гурлер синдром // Энциклопедический словарь по психологии и педагогике (рус.). — 2013.
- ↑ 1 2 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. Dermatology: 2-Volume Set. — St. Louis: Mosby, 2007. — ISBN 1-4160-2999-0. (англ.)
- ↑ Медицинская практика. Синдром Гурлер (гаргоилизм). medpractik.ru. Дата обращения 13 декабря 2014.
- ↑ Ropper AH, Samuels MA, «Chapter 37. Inherited Metabolic Diseases of the Nervous System» (Chapter). Ropper AH, Samuels MA: Adams and Victor’s Principles of Neurology, 9e: https://www.accessmedicine.com/content.aspx?aID=3636356. (англ.)
- ↑ Bonakdar-Pour, Akbar. Diagnostic Imaging of Musculoskeletal Diseases: A Systematic Approach (англ.). (англ.)
- ↑ 1 2 Scheie syndrome — National Library of Medicine — PubMed Health (англ.). ncbi.nlm.nih.gov. Дата обращения 13 декабря 2014.
- ↑ James, William D.; Berger, Timothy G.; et al. Andrews’ Diseases of the Skin: clinical Dermatology (англ.). — Saunders Elsevier, 2006. — ISBN 0-7216-2921-0. (англ.)
Литература[править | править код]
- Harrison’s Principles of Internal Medicine
Ссылки[править | править код]
- Мукополисахаридозы
- Белорусская организация больных мукополисахаридозом и другими редкими генетическими заболеваниями
- Справочник химика 21: Лизосомные болезни накопления
Источник