Мраморная болезнь код мкб

Рубрика МКБ-10: Q78.2
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q78 Другие остеохондродисплазии
Определение и общие сведения[править]
Остеопетроз
Синонимы: мраморная болезнь
Под этим названием объединяют группу наследственных заболеваний, характеризующихся разрастанием костной ткани вследствие нарушения развития/функции остеокластов.
Заболеваемость аутосомно-рецессивным остеопетрозом составляет 1:250 000 новорожденных, а заболеваемость аутосомно-доминантным остеопетрозом — 1:20 000 новорожденных.
Этиология и патогенез[править]
Причина остеопетроза — нарушение резорбции костной ткани, обусловленное функциональными дефектами остеокластов. Мутаций как минимум в 10 генах были идентифицированы в качестве каузальные, они обнаруживаются в 70% всех случаев остеопетроза.
Клинические проявления[править]
Остеопетроз у новорожденных и грудных детей — это тяжелое аутосомно-рецессивное заболевание. Во время внутриутробного развития компактное вещество костей врастает в костномозговые полости и замещает губчатое вещество. В результате угнетается костномозговое и усиливается экстрамедуллярное кроветворение. Поэтому у новорожденных и грудных детей остеопетроз проявляется в первую очередь панцитопенией, гепатоспленомегалией и увеличением лимфоузлов (см. рис. 26.6 и рис. 26.7). Кроме того, сужаются отверстия черепа и сдавливаются черепные нервы, что приводит к слепоте и тугоухости. Из-за большого веса головы нарушается моторное развитие. Больные обычно не доживают до 10 лет.
У детей старшего возраста и подростков болезнь может быть как аутосомно-доминантной, так и Х-сцепленной и протекает не так тяжело. Иногда остеопетроз выявляют случайно при рентгенологическом исследовании. В других случаях болезнь обнаруживают при обследовании по поводу патологических переломов, боли в костях, остеомиелита, поражений черепных нервов.
Остеопетроз: Диагностика[править]
Лабораторная и инструментальная диагностика. Характерны гипокальциемия, гипофосфатемия и повышение уровня ПТГ в сыворотке (вторичный гиперпаратиреоз). Всасывание кальция в кишечнике усилено, но гипокальциемия тем не менее сохраняется, поскольку ПТГ не активирует остеокласты. При рентгенографии и денситометрии костей обнаруживают их равномерное уплотнение и увеличение костной массы; компактное и губчатое вещество имеют одинаковую плотность. При гистологическом исследовании биоптатов кости выявляются толстые прослойки неминерализованного органического матрикса, иногда встречаются признаки остеомаляции (рахита). Количество остеокластов нормальное или даже увеличенное, но они малоактивны.
Антенатальная диагностика возможна, если идентифицированы мутации в семье.
Дифференциальный диагноз[править]
Дифференциальный диагноз включают флюороз, бериллиоз, отравление свинцом или висмутом, миелофиброз, болезнь Педжета (склерозирующая форма) и злокачественные новообразования (лимфома, остеокластические метастазы).
Остеопетроз: Лечение[править]
Лечение остеопетрозных состояний исключительно симптоматическое. Имеются сообщения об успешном лечении остеопетроза макрофагальным колониестимулирующим фактором, интерфероном гамма, кальцитриолом и ПТГ.
Трансплантация костного мозга HLA-идентичных доноров в настоящее время все шире применяется для лечения новорожденных и грудных детей с тяжелым остеопетрозом.
Прогноз
Тяжелые формы инфантильного остеопетроза имеют неблагоприятный прогноз, в отсутствии лечения — дети умирают в течение первых 10 лет жизни из-за недостаточности костного мозга. Продолжительность жизни при формах с началом в зрелом возрасте нормальна.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
1. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
Названия
Название: Врожденный семейный остеопетроз.
Рентгенограмма костей таза при мраморной болезни
Описание
Врожденный семейный остеопетроз. Наследственное заболевание с разными типами наследования и клинического течения, которое характеризуется нарушением процессов оссификации костей с их уплотнением и рядом сопутствующих нарушений. Симптомами этого состояния являются ломкость костной ткани (легкое развитие травматических и патологических переломов), проявления тяжелой анемии с гепато- и спленомегалией, в ряде случаев отмечаются неврологические нарушения по причине травматизации нервов, проходящих через костные каналы. Диагностика врожденного семейного остеопетроза основывается на данных рентгенологических исследований, изучения наследственного анамнеза больного, молекулярно-генетических и общих клинических анализов. Специфического лечения этого состояния не существует, в некоторых случаях значительно улучшить состояние больного может трансплантация костного мозга.
Дополнительные факты
Врожденный семейный остеопетроз (мраморная болезнь, синдром Альберс-Шенберга) – группа генетических нарушений со сходными клиническими проявлениями, заключающимся в уплотнении костной ткани с уменьшением диаметра костно-мозговых и других каналов. Одна из форм этого заболевания была впервые описана в 1904-м году немецким хирургом Г. Альберс-Шенбергом, с тех пор наиболее распространенный (аутосомно-доминантный) тип врожденного семейного остеопетроза носит его имя. Это состояние с одинаковой частотой поражает как мужчин, так и женщин, встречаемость зависит от разновидности патологии. Так, наиболее тяжелая по своим клиническим проявлениям аутосомно-рецессивная форма врожденного семейного остеопетроза имеет частоту 1:200 000-300 000, тогда как типы заболевания с аутосомно-доминантным наследованием встречаются у 1-го человека на 20 000 населения. Другое название патологии (мраморная болезнь) обусловлено рентгенологической картиной при ней (зернистость костной ткани, напоминающая мрамор и обусловленная очагами остеопетроза), а также повышенной хрупкостью, но в тоже время твердостью костей. Врожденный семейный остеопетроз является одним из наиболее изученных генетических заболеваний, сопровождающихся гиперостозом.

Изменения костей при мраморной болезни
Причины
Непосредственной причиной всех форм врожденного семейного остеопетроза является нарушение функциональной активности остеокластов – клеток, отвечающих за деструкцию элементов костной ткани. В результате этого нарушается баланс между образованием и разрушением костей, что сопровождается увеличением плотности компактного вещества, уменьшением просвета костномозговых каналов и других отверстий в костях, изменением формы метафизов. Несмотря на остеосклероз, элементы скелета при врожденном семейном остеопетрозе обладают повышенной хрупкостью, что приводит к частым травматическим и патологическим переломам, сколиозам и другим формам деформации позвоночника. Генетическая и молекулярная природа нарушения работы остеокластов неодинакова при различных формах заболевания.
Наиболее часто встречающаяся аутосомно-доминантная форма врожденного семейного остеопетроза (синдром Альберс-Шенберга) обусловлена мутацией гена CLCN7, расположенного на 16-й хромосоме. Он кодирует последовательности одной из субъединиц хлор-специфичного ионного канала, который особенно распространен на поверхности мембран остеокластов. Он участвует в процессе образования соляной кислоты, которая необходима для растворения солей кальция, входящих в состав костной ткани. В результате дефекта гена CLCN7 структура ионного канала изменяется и приводит к нарушению его функции, в результате чего выделение соляной кислоты сильно снижается, что и является причиной врожденного семейного остеопетроза.
Врожденный семейный остеопетроз с аутосомно-рецессивным типом наследования представляет собой более генетически гетерогенное состояние, оно может быть обусловлено мутациями нескольких генов. Классический тип этой патологии, по данным современной генетики, обусловлен дефектом гена TCIRG1, локализованного на 11-й хромосоме. Продуктом его экспрессии является протеин под названием «Т-клеточного иммунного регулятора», и он же представляет собой одну из субъединиц трансмембранного белка вакуолей остеокластов, проявляющего свойства АТФ-зависимого протонного насоса. Наряду с рядом хлорных каналов, он участвует в образовании соляной кислоты, которая, как и при аутосомно-доминантном варианте врожденного семейного остеопетроза необходима для деструкции костной ткани и функционирования остеокластов. Мутации гена TCIRG1 обуславливают примерно половину всех случаев данной формы заболевания.
Другие генетические дефекты намного реже приводят к развитию врожденного семейного остеопетроза с аутосомно-рецессивным типом наследования. Так, примерно у 10% таких больных выявляются мутации гена CLCN7, ассоциированного с доминантной формой патологии – но при этом характер генетического дефекта более выраженный и приводит к прекращению экспрессии белков хлор-специфичного ионного канала. Еще более редкий тип врожденного семейного остеопетроза вызывается мутацией гена OSTM1, расположенного на 6-й хромосоме – он кодирует одноименный протеин, участвующий во внутриклеточной передаче информации в остеокластах. В результате данного генетического дефекта нарушаются практически все функции этих клеток, даже при сохраненной способности синтезировать соляную кислоту. По некоторым данным, существует разновидность врожденного семейного остеопетроза, которая передается по механизму сцепленного с полом – врачи-генетики связывают ее с мутациями гена IBKG.
Симптомы
Выраженность симптомов врожденного семейного остеопетроза и тяжесть его течения находится в сильной зависимости от формы этого заболевания. Наиболее ранними и явными проявлениями обладают аутосомно-рецессивные типы этой патологии, которые проявляются сразу при рождении, а в некоторых случаях и внутриутробно на заключительных этапах вынашивания ребенка. Ведущим проявлением на этом этапе развития врожденного семейного остеопетроза является тяжелейшая анемия, вызванная дефицитом красного костного мозга из-за сужения костномозговых каналов. Она приводит к многочисленным вторичным нарушениям – гепато- и спленомегалии, имеющие прогрессирующий характер, бледность кожных покровов, признаки кислородного голодания тканей. Нередко у страдающих врожденным семейным остеопетрозом младенцев определяют гидроцефалию, в дальнейшем многие больные теряют слух и зрение из-за сужения отверстий черепа, через которые проходят соответствующие нервы. Лицо таких больных имеет характерные особенности – широко расставленные глаза, вдавленная переносица, толстые и диспропорциональные губы. Многочисленные нарушения при аутосомно-рецессивной форме врожденного семейного остеопетроза имеют тенденцию к прогрессированию, что нередко приводит к смерти больных до достижения ими 3-8 лет.
Диагностика
Для диагностики врожденного семейного остеопетроза применяют рентгенологические методики, общие анализы крови, молекулярно-генетические исследования, изучение наследственного анамнеза больного. На рентгенограммах выявляют общее уплотнение костной ткани (диффузный остеосклероз), выраженность которого больше при аутосомно-рецессивных вариантах заболевания. В некоторых элементах скелета (фаланги пальцев, подвздошные кости, позвонки) остеопетроз имеет очаговый характер, создавая характерную рентгенологическую картину, известную как «кость в кости». В длинных трубчатых костях изменяется форма метафизов и размеры костномозгового канала – от незначительного уменьшения при доброкачественном типе врожденного семейного остеопетроза до практически полного исчезновения при рецессивном. На рентгенограммах также могут выявляться признаки гидроцефалии, искривления позвоночника, у взрослых – следы многочисленных заживших переломов.
В анализах крови определяется резкое снижение уровня эритроцитов и гемоглобина, нередко снижаются показатели относительно других клеточных элементов. У детей с рецессивной формой врожденного семейного остеопетроза это может иметь жизнеугрожающий характер, тогда как у взрослых (аутосомно-доминантная форма) анемия выражена слабо или вообще не обнаруживается. Молекулярно-генетические исследования наиболее часто сводятся к прямому секвенированию гена TCIRG1, мутации которого приводят к злокачественному типу этого заболевания. Поводом для такого анализа служат, помимо характерной клинической картины, наличие подобных проявлений у родственников родителей больного – это может указывать на аутосомно-рецессивный характер передачи болезни.
Лечение
Специфическое лечение аутосомно-рецессивных форм врожденного семейного остеопетроза (как и доминантных) не разработано – некоторые исследователи в качестве такового указывают на трансплантацию костного мозга. Так как некоторые остеокласты являются вариантом тканевых макрофагов (то есть, имеют костномозговое происхождение) такой метод лечения способен улучшить состояние больного за счет «замены» собственных клеток донорскими остеокластами без генетических дефектов. Действительно, в ряде случаев врожденного семейного остеопетроза трансплантация костного мозга приводила к улучшению, но примеров полного выздоровления зафиксировано не было. Остальные терапевтические мероприятия имеют поддерживающий и симптоматический характер – применение антианемических средств (препаратов железа, эритропоэтинов), витамина Д, лечебная физкультура и гимнастика. При аутосомно-доминантной форме врожденного семейного остеопетроза заметное улучшение наблюдается при санаторно-курортном лечении.
Источник
Исключены:
- лихорадка неясного происхождения (во время) (у):
- родов (O75.2)
- новорожденного (P81.9)
- лихорадка послеродового периода БДУ (O86.4)
Боль в области лица
Исключены:
- атипичная боль в области лица (G50.1)
- мигрень и другие синдромы головной боли (G43-G44)
- невралгия тройничного нерва (G50.0)
Включена: боль, которая не может быть отнесена к какому-либо определенному органу или части тела
Исключены:
- хронический болевой личностный синдром (F62.8)
- головная боль (R51)
- боль (в):
- животе (R10.-)
- спине (M54.9)
- молочной железе (N64.4)
- груди (R07.1-R07.4)
- ухе (H92.0)
- области таза (H57.1)
- суставе (M25.5)
- конечности (M79.6)
- поясничном отделе (M54.5)
- области таза и промежности (R10.2)
- психогенная (F45.4)
- плече (M25.5)
- позвоночнике (M54.-)
- горле (R07.0)
- языке (K14.6)
- зубная (K08.8)
- почечная колика (N23)
последние изменения: январь 2015
R53
Недомогание и утомляемость
Астения БДУ
Слабость:
- БДУ
- хроническая
Общее физическое истощение
Летаргия
Усталость
Исключены:
- слабость:
- врожденная (P96.9)
- старческая (R54)
- истощение и усталость (вследствие) (при):
- нервной демобилизации (F43.0)
- чрезмерного напряжения (T73.3)
- опасности (T73.2)
- теплового воздействия (T67.-)
- неврастении (F48.0)
- беременности (O26.8)
- старческой астении (R54)
- синдром усталости (F48.0)
- после перенесенного вирусного заболевания (G93.3)
последние изменения: январь 2012
Старческий возраст без упоминания о психозе
Старость без упоминания о психозе
Старческая:
- астения
- слабость
Исключен: старческий психоз (F03)
R55
Обморок [синкопе] и коллапс
Кратковременная потеря сознания и зрения
Потеря сознания
Исключены:
- нейроциркуляторная астения (F45.3)
- ортостатическая гипотензия (I95.1)
- неврогенная (G23.8)
- шок:
- БДУ (R57.9)
- кардиогенный (R57.0)
- осложняющий или сопровождающий:
- аборт, внематочную или молярную беременность (O00-O07, O08.3)
- роды и родоразрешение (O75.1)
- послеоперационный (T81.1)
- приступ Стокса-Адамса (I45.9)
- обморок:
- синокаротидный (G90.0)
- тепловой (T67.1)
- психогенный (F48.8)
- бессознательное состояние БДУ (R40.2)
последние изменения: январь 2016
Исключены: судороги и пароксизмальные приступы (при):
- диссоциативные (F44.5)
- эпилепсии (G40-G41)
- новорожденного (P90)
Исключены:
- шок (вызванный):
- анестезией (T88.2)
- анафилактический (вследствие):
- БДУ (T78.2)
- неблагоприятной реакции на пищевые продукты (T78.0)
- сывороточный (T80.5)
- осложняющий или сопровождающий аборт, внематочную или молярную беременность (O00-O07, O08.3)
- воздействием электрического тока (T75.4)
- в результате поражения молнией (T75.0)
- акушерский (O75.1)
- послеоперационный (T81.1)
- психический (F43.0)
- травматический (T79.4)
- синдром токсического шока (A48.3)
последние изменения: январь 2015
R58
Кровотечение, не классифицированное в других рубриках
Кровотечение БДУ
Включены: опухшие железы
Исключены: лимфаденит:
- БДУ (I88.9)
- острый (L04.-)
- хронический (I88.1)
- мезентериальный (острый) (хронический) (I88.0)
Исключена: задержка полового созревания (E30.0)
Исключены:
- булимия БДУ (F50.2)
- расстройства приема пищи неорганического происхождения (F50.-)
- недостаточность питания (E40-E46)
Исключены:
- синдром истощения как результат заболевания, вызванного ВИЧ (B22.2)
- злокачественная кахексия (C80.-)
- алиментарный маразм (E41)
последние изменения: январь 2010
Эта категория не должна использоваться в первичном кодировании. Категория предназначена для использования в множественном кодировании, чтобы определить данный синдром, возникший по любой причине. Первым должен быть присвоен код из другой главы, чтобы указать причину или основное заболевание.
добавлено: январь 2010
R69
Неизвестные и неуточненные причины заболевания
Болезненность БДУ
Недиагностированная болезнь без уточнения локализации или пораженной системы
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Периодическая болезнь.
Периодическая болезнь
Описание
Периодическая болезнь. Генетическая патология, характеризующая нарушением регуляции воспалительных процессов, особенно в области серозных (брюшины, плевры) и синовиальных оболочек. Проявления этого заболевания различны, чаще всего регистрируются боли в животе (картина острого перитонита), нарушения со стороны плевральной полости, приступы лихорадки, болезненность и отечность суставов. Диагностика производится на основании клинической картины, изучения наследственного анамнеза и молекулярно-генетических анализов, вспомогательную роль играет определение национальности больного. Лечение периодической болезни только симптоматическое и поддерживающее, специфической терапии на сегодняшний момент не существует.
Дополнительные факты
Периодическая болезнь (средиземноморская семейная лихорадка) – наследственное заболевание, причиной которого являются нарушения в регуляции воспалительного и иммунного ответа на уровне гранулоцитов. Впервые данная патология была описана в 1948-м году американским врачом Райманном, который из-за повторяющихся тяжелых приступов дал ей название «периодическая болезнь». С первых лет изучения была выявлена главная особенность этой патологии – она возникает только у уроженцев средиземноморского региона и Малой Азии, главным образом у армян, арабов, греков, испанцев, итальянцев, евреев-сефардов и турок. У представителей иных национальностей отмечены лишь спорадические и статистические незначимые случаи периодической болезни. Поэтому фактор национальности больного и его предков играет немаловажную роль в диагностике данного состояния. Встречаемость периодической болезни у различных этносов средиземноморского региона отличается, она наиболее высока у турок, арабов и армян, несколько ниже у евреев-сефардов, еще реже это заболевание встречается у греков, итальянцев и испанцев. По некоторым данным, носительство патологического гена в определенных регионах затрагивает 20% населения, а заболеваемость составляет 1:1000-2500. Периодическая болезнь наследуется по аутосомно-рецессивному механизму и с одинаковой частотой поражает как мальчиков, так и девочек.
Периодическая болезнь
Причины
Долгое время этиология и патогенез периодической болезни оставались неизвестными, лишь достижения современной генетики позволили больше узнать об этом заболевании. Наиболее часто причиной данной патологии являются мутации гена MEFV, расположенного на 16-й хромосоме. Ген кодирует белок под названием маренострин (другое название – пирин), который выполняет функции одного из центральных регуляторов воспалительной реакции и первичного иммунного ответа. Маренострин тормозит дегрануляцию нейтрофилов и угнетает их адгезивные свойства, тем самым ослабляя и ингибируя чрезмерную реакцию иммунной системы. При периодической болезни миссенс-мутации гена MEFV приводят к изменению структуры маренострина, тем самым нарушая его функции. Это снижает порог дегрануляции нейтрофилов, что облегчает развитие острых воспалительных реакций и формирует клиническую картину периодической болезни.
Кроме того, дефекты маренострина приводят к каскадным патологическим реакциям в иммунной системе и организме в целом. Значительно уменьшается активность ингибитора одного из компонентов системы комплемента – С5а. Последний постепенно накапливается в серозных оболочках, а при достижении высоких концентраций провоцирует бурную воспалительную реакцию. Это обстоятельство объясняет определенные свойства периодической болезни – преимущественное поражение серозных оболочек, а также сезонность заболевания (для накопления достаточных концентраций С5а необходимо несколько месяцев). В некоторых случаях для периодической болезни характерно также раннее развитие амилоидоза, однако его патогенез остается неясным.
Все вышеперечисленные процессы возникают при наличии у человека двух аллелей дефектного гена MEFV, то есть у гомозигот, так как периодическая болезнь является аутосомно-рецессивным заболеванием. Существует теория, согласно которой гетерозиготы из-за снижения ингибирования адгезивных свойств гранулоцитов обладают повышенной резистентностью к бактериальным инфекциям. Отчасти это может объяснить столь высокую встречаемость патологической формы гена и его носительства среди этносов средиземноморского региона. Кроме того, существуют указания, что некоторые формы периодической болезни обусловлены дефектом генов на 19-й хромосоме, однако точно идентифицировать их пока не удалось.
Симптомы
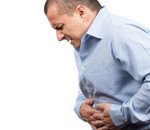
Клиническая картина периодической болезни отличается большим разнообразием, однако причины этого пока достоверно неизвестны – предполагается взаимосвязь между отдельными типами мутаций и формами заболевания. Удалось выяснить, что, например, амилоидоз, в среднем поражающий 30-35% больных, намного чаще возникает у арабов и турок, нежели у армян. Постоянным симптомом периодической болезни (наблюдается в 99% случаев) является выраженная лихорадка, которая не купируется традиционными жаропонижающими средствами и антибиотиками. В зависимости от клинической формы заболевания повышение температуры тела может сочетаться с другими проявлениями. На сегодняшний день выделяют четыре основные клинические формы периодической болезни: абдоминальную, торакальную, суставную и псевдомалярийную.
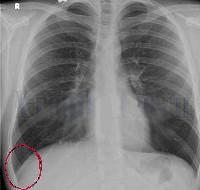
Абдоминальная форма периодической болезни характеризуется типичной картиной «острого живота» при перитоните, включает в себя резкое повышение температуры тела до 40-41 градуса, опоясывающие боли, ригидность мышц брюшной стенки, тошноту и рвоту. Такие проявления сохраняются на протяжении нескольких суток, после чего постепенно стихают. За это время более чем у половины больных периодической болезнью ошибочно диагностируется гнойный перитонит (на самом деле при данной патологии развивается асептическое воспаление брюшины), аппендицит, прободная язва желудка, производятся ненужные хирургические операции. Торакальная форма периодической болезни создает картину выпотного плеврита, который может иметь одно- или двухсторонний характер. Это приводит к болям в грудной клетке, затрудненному дыханию, одышке и другим типичным проявлениям гнойного или экссудативного плеврита, что также нередко становится причиной постановки ошибочного диагноза. Проявления торакальной формы периодической болезни постепенно стихают на протяжении 7-10 дней.
Суставная форма периодической болезни характеризуется развитием отеков и болезненности нескольких (реже – одного) суставов, резким покраснением кожи на пораженной области. Симптомы сохраняются на протяжении 2-4 недель, артралгия может наблюдаться в течение нескольких месяцев. При этом каких-либо постоянных нарушений в суставах (ограничение подвижности, контрактуры) при периодической болезни не возникает. Псевдомалярийная форма заболевания характеризуется приступами сильной лихорадки длительностью 3-7 дней, после чего температура тела больного приходит в норму. Никаких проявлений со стороны других органов на начальных этапах развития патологии при этом не определяется.
Одышка. Рвота. Тошнота.
Классификация
По данным медицинской статистики, изолированные клинические формы (абдоминальная, торакальная и другие) имеют место примерно в 20% случаев периодической болезни. Намного чаще встречается сочетание нескольких клинических типов патологии (торакальной и суставной, лихорадки на фоне абдоминальных симптомов). При отсутствии лечения периодической болезни примерно у трети больных развивается амилоидоз почек, который приводит к хронической почечной недостаточности и уремии. В 20% случаев на фоне вышеперечисленных проявлений могут отмечаться дерматологические симптомы: папулезная сыпь, крапивница, рожеподобное воспаление. Редко при периодической болезни развиваются асептические менингиты и перикардиты, а также воспаление яичек (орхит).
Диагностика
В ряде случаев диагностика периодической болезни может быть сопряжена со значительными сложностями по причине выраженности, и, в то же время, неспецифичности ее проявлений. Эта особенность заболевания может стать причиной диагностических ошибок с далеко идущими последствиями – так, при картине «острого живота» больным часто производят ненужные операции, при асептических плевритах и менингитах назначают высокие дозы антибиотиков. В случае артралгии и постановки неправильного диагноза (например, ревматоидный артрит) больному периодической болезнью могут назначать сильнодействующие иммуносупрессивные средства. Поэтому при наличии таких симптомов у пациентов, являющихся уроженцами средиземноморского региона, обязательно следует учитывать возможность наличия этого генетического заболевания.
В процессе диагностики периодической болезни используют данные изучения наследственного анамнеза больных и молекулярно-генетических анализов. Как правило, наследственный анамнез у таких пациентов отягощен (крайне редко встречаются спорадические формы), подобные проявления выявляются у предков или родственников. Окончательно подтвердить или опровергнуть наличие периодической болезни может врач-генетик посредством генетического исследования. Существует распространенная методика поиска наиболее часто встречающихся при этом заболевании мутаций гена MEFV – M694V и V726A, которые обуславливают более 75% всех случаев данной патологии. Однако более редкие дефекты MEFV могут остаться незамеченными – для их определения применяют секвенирование всей последовательности гена.
Лечение
Лечение периодической болезни в основном симптоматическое. При выраженных болях в животе, груди, суставах применяют нестероидные противовоспалительные средства и другие анальгетики, в редких случаях (при болях, сопровождающих абдоминальную форму болезни) могут назначаться наркотические обезболивающие препараты. Гидроторакс при плеврите устраняют пункцией и назначением диуретических средств. Для профилактики приступов, снижения выраженности симптомов и общего улучшения состояния пациентов назначают длительный прием колхицина. При развитии почечной недостаточности из-за амилоидоза больным периодической болезнью рекомендуется регулярный гемодиализ.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник