Мозжечковая атаксия мари код мкб

Рубрика МКБ-10: G11.3
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G10-G13 Системные атрофии, поражающие преимущественно центральную нервную систему / G11 Наследственная атаксия
Определение и общие сведения[править]
Атаксия-телеангиэктазия
Синонимы: синдром Луи-Бар
Определение и общие сведения
Атаксия-телеангиэктазия проявляется тяжелым комбинированным иммунодефицитом (поражающим в основном гуморальный иммунный ответ) и прогрессирующей мозжечковой атаксией. Синдром Луи-Бар характеризуется неврологическими симптомами, телеангиэктазиями, повышенной восприимчивость к инфекциям и более высоким риском развития новообразований.
Средняя распространенность оценивается порядка 1/100 000 детей.
Этиология и патогенез[править]
Синдром Луи-Бар является аутосомно-рецессивным заболеванием, вызывается инактивирующей мутацией гена ATM (11q22.3). Этот ген экспрессируется повсеместно и кодирует киназу протеина, который играет ключевую роль в контроле двухнитевого разрыва (DSB) процесса репарации ДНК, в частности, в клетках Пуркинье мозжечка и головного мозга, эндотелиальных клетках кожи и конъюнктивы. Атаксия-телеангиэктазия-подобное расстройство представляет собой редкий вариант синдрома Луи-Бар вызвано инактивации гена MRE11 (11q21), который также кодирует белок, участвующий в двухнитевом разрыве репарации ДНК.
Клинические проявления[править]
Тяжесть неврологических, иммунных и легочных проявлений синдрома широко варьирует между пациентами. Манифестация заболевания обычно происходит возрасте от 1 до 2 лет аномальными движениями головы, потерями равновесия, а затем появлением невнятной речи и аномальных движений глаз. Плохая координация и тремор конечностей могут появиться ближе к 9-10 годам жизни и постепенно прогрессирует. Хореоатетоз является довольно распространенным симптомом атаксии-телеангиэктазии. В большинстве случаев, интеллект не страдает, но около 30% пациентов испытывают трудности в обучении или демонстрируют умственной недостаточности умеренной степени выраженности. Кожномукозные телеангиэктазии появляются от в возрасте от 3 до 6 лет или в подростковом возрасте. Иммунодефицит вызывает рецидивирующие синуситы и инфекций дыхательных путей, последние могут сопровожлаться бронхоэктазами. Довольно часто наблюдается задержка роста ребенка.
Телеангиэктазии у больных атаксией-телеангиэктазией обычно локализуются на конъюнктиве склеры, а также на коже лица, особенно выражены они под глазами и в области ушных раковин. Возможны они на шее, туловище, конечностях. Цвет их обычно темно-красный. Возможны и выраженные локальные расширения сосудов мозговых оболочек и вещества мозга.
Мозжечковая атаксия с нарушением репарации ДНК: Диагностика[править]
Установление клинического диагноза на ранней стадии течения заболевания является сложной задачей, но увеличение уровня альфа-фетопротеина в сыворотке крови и цитогенетический анализ могут помочь подтвердить диагноз (7; 14 транслокации).
Сосудистые изменения в конъюнктиве склеры глазных яблок могут рассматриваться при атаксии-телеангиэктазии (синдром Луи-Бар) как надежный диагностический тест. При офтальмоскопии на глазном дне могут быть обнаружены ангиомы, кисты, глиальные разрастания в дисках зрительных нервов или признаки застоя в их венах. Острота зрения у большинства больных сохраняется в пределах от 0,3 до 1,0.
Дифференциальный диагноз[править]
Дифференциальный диагноз должен включать атаксию-глазодвигательные апраксию типы 1 и 2. Пренатальный диагноз возможен при идентифиции инактивирующей мутации АТМ гена.
Мозжечковая атаксия с нарушением репарации ДНК: Лечение[править]
Терапия является симптоматической и включает в себя физиотерапию, занятия с логопедом и лечение инфекций и легочных осложнений. Бета-блокаторы могут уменьшить тремор и улучшить производительность тонких движений.
Пациенты часто нуждаются в использовании инвалидной коляски с возраста 10-11 лет. Прогноз является серьезным, из-за частых респираторных инфекций, нейродегенеративных растройств, ускоренного старения кожи и подкожной клетчатки и повышенного риск развития новообразований (у 35% пациентов развивается рак в возрасте до 20 лет ).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Офтальмоневрология [Электронный ресурс] / А. С. Никифоров, М. Р. Гусева — М. : ГЭОТАР-Медиа, 2014.
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
- Описание
- Симптомы (признаки)
- Лечение
Краткое описание
Атаксия — двигательное расстройство, проявляющееся в неспособности к координации произвольных движений; может быть следствием мозжечковых нарушений, расстройств двигательной или чувствительной систем.
Код по международной классификации болезней МКБ-10:
- G11 Наследственная атаксия
- G60.2 Невропатия в сочетании с наследственной атаксией
- G80.4 Атаксический церебральный паралич
- R26 Нарушения походки и подвижности
- R27.0 Атаксия неуточненная
Генетические аспекты. В основе ряда форм находятся дефекты генов кальциевых и других каналов. Типы и гены: Атаксия спиномозжечковая: • тип 1: 164400, ATX1, SCA1 (атаксин 1, 601556, 6p23); • тип 2: 183090, ATX2, SCA2 (атаксин 2, 601517, 12q24); • тип 4: SCA4, 600223, 16q22.1; • тип 5: SCA5, 600224, 11p11 q11; • тип 6: 183086, CACNA1A, CACNL1A4, SCA6 (a1A СЕ кальциевого потенциалзависимого канала P/Q типа, 601011, 19p13); • тип 7: 164500, SCA7, OPCA3, 3p21.1 p12; • тип 8 (инфантильная с сенсорной невропатией): 271245, SCA8, IOSCA, 10q24. Атаксия с недостаточностью витамина Е: 277460, TTPA, TTP1, AVED (600415 [токоферол транспортирующий белок], 8q13.1 q13.3). Атаксия мозжечковая: • Каймановых островов: 601238, ATCAY, CLAC, 19p13.3.
Симптомы (признаки)
Классификация и клиническая картина
• По характеру •• Статическая (атаксия туловища) — нарушение равновесия в положении стоя и сидя •• Динамическая (локомоторная) — нарушение координации при произвольных движениях конечностей, особенно верхних •• Статико — локомоторная — расстройство стояния и ходьбы.
• По локализации основного поражения •• Сенситивная (заднестолбовая) атаксия возникает при поражении задних столбов спинного мозга, реже — при поражении периферических нервов, задних корешков, зрительного бугра, коры теменной области. Наблюдают при нейросифилисе (табетическая атаксия, часто сочетается с синдромом Аргайлла Робертсона), фуникулярном миелозе, некоторых формах полиневропатий, сосудистых нарушениях, опухолях. Характерна общая неустойчивость. При ходьбе больной чрезмерно сгибает ноги в коленных и тазобедренных суставах и с излишней силой опускает их на пол (штампующая походка). Контроль зрения уменьшает, а закрывание глаз резко усиливает явления атаксии •• Мозжечковая атаксия — статическая или динамическая атаксия, возникающая при поражении мозжечка и/или его проводящих путей. Наблюдают при рассеянном склерозе, опухолях, энцефалитах, сосудистых очагах в мозжечке и стволе мозга, дегенеративных заболеваниях мозжечка ••• В позе Ромберга и при ходьбе пациент отклоняется или падает в сторону поражённого полушария мозжечка ••• При поражении червя мозжечка наблюдают падение в разные стороны, нередко назад (пьяная походка) ••• Контроль зрения мало влияет на выраженность нарушений координации ••• Речь замедленная, растянутая, толчкообразная, иногда скандированная (артикуляционная атаксия) ••• Изменения почерка — неровность, размашистость, макрография ••• На стороне поражения обычно снижен мышечный тонус •• Вестибулярная атаксия возникает при поражении вестибулярного аппарата. Развивается при болезни Меньера и других заболевания внутреннего уха, стволовых энцефалитах, опухолях IV желудочка. Характерные признаки: системное головокружение (больному кажется, что все предметы движутся в определённом направлении); горизонтальный нистагм, тошнота, рвота, усиливающиеся при поворотах головы; больной беспорядочно шатается в стороны или падает •• Корковая атаксия возникает при поражении лобной или височно — затылочной коры. Регистрируют при опухолях, абсцессах, нарушении мозгового кровообращения ••• Максимально страдает контралатеральная очагу нога, появляются неустойчивость при ходьбе, особенно на поворотах, отклонение в сторону, противоположную поражённому полушарию. Контроль зрения мало влияет на степень атаксии. В случае тяжёлых поражений больной совсем не может стоять и ходить (астазия — абазия) ••• Другие симптомы поражения лобной доли (изменение психики, хватательный рефлекс, нарушение обоняния).
• По этиологии дополнительно выделяют •• Алкогольная атаксия, возникает при алкогольной интоксикации •• Истерическая атаксия — заболевание психогенного происхождения с большой вариабельностью проявлений, зависящей от эмоционального состояния больного.
Лечение
Лечение • Симптоматическое • Лечебная гимнастика, направленная на тренировку чувства равновесия.
МКБ-10 • G11 Наследственная атаксия • G60.2 Невропатия в сочетании с наследственной атаксией • G80.4 Атаксический церебральный паралич • R26 Нарушения походки и подвижности • R27.0 Атаксия неуточнённая
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Лечение
Названия
Название: Спиноцеребеллярные атаксии.
Спиноцеребеллярные атаксии
Описание
Спиноцеребеллярные атаксии. Группа генетически разнородных наследственных заболеваний неврологического характера, которые проявляются различными расстройствами работы мозжечка и иногда базальных ядер головного мозга. Симптомами этого состояния являются: развитие атаксии и неустойчивой походки, нарушение координации движений и другие неврологические проявления. Диагностика спиноцеребеллярных атаксий производится на основании данных неврологического осмотра, изучения наследственного анамнеза больного, магнитно-резонансной томографии и молекулярно-генетических исследований. Специфического лечения этой патологии на сегодняшний момент не существует, для сохранения оптимального качества жизни больного используют методы поддерживающей и симптоматической терапии.
Дополнительные факты
Спиноцеребеллярные атаксии – группа наследственных неврологических состояний, характеризующихся развитием прогрессирующей дегенерации клеток мозжечка и иногда базальных ядер вплоть до их полной атрофии. Впервые одно из заболеваний этой группы было описано еще в 1891 году немецким невропатологом П. Менцелем, который выявил развитие атаксии, офтальмоплегии и других неврологических нарушений в рамках одной семьи. Дальнейшие исследования показали, что это состояние (известное сейчас как спиноцеребеллярная атаксия 1-го типа) наследуется по аутосомно-доминантному механизму.
В настоящий момент методами современной генетики удалось обнаружить более 20 различных генетических вариантов этого заболевания, при этом более 90% всех случаев обуславливает только 6 из них (1, 2, 3, 6, 7 и 8-й типы). Все формы спиноцеребеллярных атаксий характеризуются аутосомно-доминантным наследованием с явлениями антиципации (усиления выраженности патологии от поколения к поколению) и «отцовской передачи» – более яркой клинической картине заболевания при его наследовании от отца. Поэтому в ряде регионов в общей структуре патологии наблюдается незначительное превалирование больных мужского пола. Общая встречаемость спиноцеребеллярной атаксии колеблется в широких пределах (1-24:100 000), при этом 1-й тип распространен в России и большей части Европы, 2-й – в Индии, 3-й – в Германии и Японии.
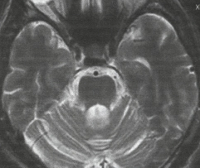
Спиноцеребеллярные атаксии
Причины
Несмотря на значительное генетическое и отчасти клиническое разнообразие спиноцеребеллярных атаксий, молекулярные механизмы генетических нарушений при этих заболеваниях очень сходны. Основная причина патологии заключается в изменении количества тринуклеотидных последовательностей (CAG) в кодирующей части ассоциированных с заболеванием генов. Это приводит к увеличению количества аминокислоты глутамина в полученном белке, что изменяет физико-химические свойства протеина и нарушает его функции. В ряде случаев вышеуказанные белки прямо или косвенно участвуют в метаболизме нервной ткани, поэтому изменение их структуры приводит к спиноцеребеллярной атаксии. В настоящее время лучше всего изучены молекулярные механизмы 6 основных разновидностей этого заболевания – данные формы патологии встречаются наиболее часто и в совокупности составляют более 90% случаев спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 1. Го типа считается самым распространенным и самым изученным вариантом данной патологии. Ее причиной выступают мутации в гене ATXN1, который располагается на 6-й хромосоме. В норме данный ген имеет не более 36 CAG-повторов, увеличение их количества приводит к развитию заболевания. Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий белок, активно участвующий в метаболизме клеток Пуркинье мозжечка – при наличии мутантной разновидности гена это приводит к появлению агрегантов и постепенной дегенерации, что и становится причиной спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 2. Го типа — менее распространенный вариант заболевания, этиология не так тщательно изучена. Причиной патологии является увеличение количества CAG-повторов в гене ATXN2, локализованном на 12-й хромосоме. В здоровом варианте гена количество вышеуказанных последовательностей составляет от 15 до 36, тогда как при спиноцеребеллярной атаксии их может быть свыше 100. Функции белка, который кодируется геном ATXN2, на сегодняшний момент неизвестны.
Спиноцеребеллярная атаксия тип 3 (другое название – болезнь Мачадо-Джозефа в честь двух больных, у которых впервые было описано данное состояние) – причиной этого варианта патологии выступают нарушения в гене ATXN3, расположенном на 14-й хромосоме. В норме количество CAG-повторов в этом гене не превышает 47, при развитии заболевания обнаруживается от 53 до 68 повторов. Данный ген кодирует белок, который предположительно участвует в энергетическом обмене нейронов мозжечка и базальных ядер.
Спиноцеребеллярная атаксия тип 6. Сравнительно редкий вид заболевания, обусловленный дефектами в гене CACNA1A, локализованном на 19 — й хромосоме. Для развития патологии достаточно очень незначительного увеличения количества CAG-повторов – если в нормальном варианте гена их обнаруживают 5-20, то при наличии атаксии – 21-26. Ген CACNA1A кодирует белок-субъединицу кальциевых каналов, расположенных на нейронах мозжечка. Помимо спиноцеребеллярной атаксии, нарушения в гене CACNA1A обуславливают развитие эпизодической атаксии и некоторые наследственные формы мигрени.
Спиноцеребеллярная атаксия тип 7. Данная разновидность патологии вызывается нарушениями структуры гена ATXN7, который располагается на 3 — й хромосоме. У здорового человека количество CAG-повторов составляет не более 35, тогда как при заболевании их количество может достигать нескольких сотен. Функции белка, который кодирует ген ATXN7, на сегодняшний момент изучаются.
Спиноцеребеллярная атаксия тип 8 обусловлена генетическим дефектом гена ATXN8, расположенного на 13-й хромосоме. Как и в других случаях, суть генетического дефекта при этом состоянии заключается в изменении количества тринуклеотидных последовательностей CAG – обычно их около 15-50, тогда как при патологии количество повторов может составлять свыше 1200.
Практически при любом типе спиноцеребеллярной атаксии патологическая форма белка, чрезмерно богатая глутамином, формирует отложения в ядрах или цитоплазме нейронов мозжечка и базальных ядер в виде плотных агрегатов. Этот процесс идет тем быстрее, чем сильнее количество CAG-повторов в ключевом гене отличается от нормы. Этим же объясняется механизм антиципации симптомов спиноцеребеллярной атаксии – в процессе мейоза при образовании половых клеток количество вышеуказанных тринуклеотидных последовательностей может увеличиваться, что приводит к усилению симптомов.
Классификация
Так как подобное явление чаще имеет место при формировании мужских половых клеток, это становится причиной так называемой «отцовской передачи», когда антиципация регистрируется только при передаче заболевания от отца потомству. Многие врачи-генетики полагают, что основная причина спиноцеребеллярных атаксий лежит не в увеличении «гистидиновых» тринуклеотидов, а в делеции так называемых регулирующих триплетов, разделяющих участки CAG-повторов. Например, при первом типе заболевания это CAT, при втором CAA – они регулируют количество CAG-повторов и сохраняют стабильность их количества во время мейоза.
Симптомы
Несмотря на значительное генетическое разнообразие спиноцеребеллярных атаксий, проявления разных типов этого заболевания в целом сходны и различаются только второстепенными деталями – возрастом манифестации, особенностями некоторых симптомов. Практически все формы патологии не регистрируются в детском возрасте – лишь отдельные случаи 1 и 2-го типов были замечены у детей младше 7 лет, средний возраст их манифестации – 18-30 лет. Спиноцеребеллярные атаксии 3, 6 и 7-го типов характеризуются еще более поздним развитием – их манифестация практически всегда происходит у лиц старше 30 лет. Нередко подобные нарушения выявляются и у пожилых людей, что затрудняет дифференциальную диагностику этого состояния с болезнью Паркинсона и другими нейродегенеративными заболеваниями старшего возраста.
Чаще всего развитие спиноцеребеллярной атаксии начинается с появления простой неуклюжести в движениях, особенно при ходьбе, беге. В дальнейшем возникает тремор рук, нарушения походки, паралич глазодвигательных мышц (офтальмоплегия), изменяется почерк больного (становится крупнее, строки неровные). В конечном итоге заболевание приводит к выраженной мозжечковой атаксии, расстройствам пирамидальных и экстрапирамидальных путей, паркинсонизму. Некоторые формы патологии характеризуются выраженными нарушениями зрения – развитием атрофии зрительного нерва, пигментной дегенерации сетчатки и других процессов.
Тремор.
Лечение
Выявление спиноцеребеллярной атаксии производится на основании данных неврологического осмотра, изучения наследственного анамнеза, магнитно-резонансной томографии головного мозга и молекулярно-генетических исследований. При осмотре больных на разных стадиях развития патологии определяются различные по выраженности неврологические нарушения – тремор конечностей, атаксия, изменения речи и голоса, на конечных этапах – дисфагия. Некоторые формы спиноцеребеллярной атаксии сопровождаются достаточно быстрым развитием нарушений зрения, приводящим к полной слепоте. Многолетнее наблюдение за такими больными подтверждает неуклонно прогрессирующее течение заболевания. При изучении наследственного анамнеза могут определяться характерные признаки спиноцеребеллярной атаксии – аутосомно-доминантное наследование, наличие антиципации при передаче болезни от отца.
На МРТ головного мозга при спиноцеребеллярной атаксии обнаруживаются очаги демиелинизации и нейродегенерации в области полушарий, червя мозжечка и базальных ядер. На терминальных стадиях развития заболевания может отмечаться полная атрофия мозжечка. Молекулярно-генетические исследования при спиноцеребеллярной атаксии сводятся к поиску патологически увеличенного количества CAG-повторов в генах, ассоциированных с этим заболеванием. В настоящее время большинство лабораторий мира осуществляет поиск этого дефекта в генах, наиболее часто приводящих к развитию патологии – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 и CACNA1A.
Специфическое лечение патологии отсутствует, поддерживающая терапия способна несколько замедлить развитие спиноцеребеллярной атаксии, но единого мнения по поводу ее эффективности на сегодняшний момент нет. Применяют витаминотерапию (Е, А, группы В), ноотропные средства, стимуляторы обмена веществ (рибоксин) и метаболизма в нервной ткани. При развитии непроизвольных движений рекомендуют использовать клоназепам и галоперидол. Важную роль в сдерживании прогрессирования спиноцеребеллярной атаксии играет лечебная физкультура – регулярное выполнение правильно подобранного комплекса упражнений позволяет укрепить мышцы и снизить выраженность расстройств равновесия. С этой же целью рекомендуют проведение сеансов лечебного массажа, процедуры электромиостимуляции.
Источник