Миопатия эрба рота код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Прогрессирующая мышечная дистрофия Эрба-Рота.
Прогрессирующая мышечная дистрофия Эрба-Рота
Описание
Прогрессирующая мышечная дистрофия Эрба. Рота — аутосомно — рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. Е. Начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений. Диагностика осуществляется методами неврологического осмотра, электрофизиологического тестирования, генеалогического исследования и генетического анализа, по показаниям проводится гистологический анализ биоптата мышечной ткани. Лечение только симптоматическое, позволяющее лишь продлить двигательную активность пациентов. Итог заболевания — полная обездвиженность.
Дополнительные факты
Прогрессирующая мышечная дистрофия Эрба-Рота — самый полиморфный вариант наследственной миодистрофии. От других видов прогрессирующей мышечной дистрофии вариант Эрба-Рота отличается вариабельностью как времени дебюта заболевания, так его клинической картины и течения. Заболевание описано в 1882 г. Немецким неврологом Вильгельмом Эрбом. Одновременно в России изучением этой патологии занимался В. К. Рот, для обозначения миодистрофии он ввел термин «мышечная сухотка». В современной мировой и отечественной неврологии в честь этих исследователей употребляется название «прогрессирующая мышечная дистрофия Эрба-Рота». Частота встречаемости заболевания находится в пределах от 1,5 до 2,5 случаев на 100 тыс. Населения.
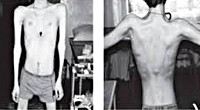
Прогрессирующая мышечная дистрофия Эрба-Рота
Причины
Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов. На сегодняшний день генетике известно не менее 9 хромосомных локусов, аберрации в которых приводят к развитию миодистрофии Эрба-Рота. Чаще всего наблюдаются мутации в локусах 15q15-q21. 1, 13q.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Симптомы
Прогрессирующая мышечная дистрофия Эрба-Рота манифестирует в среднем в возрасте 13-16 лет. Однако известны отдельные случаи дебюта болезни в раннем детском возрасте и в возрасте после 20 лет. Мышечная слабость и атрофии возникают в первую очередь в мускулатуре тазового пояса и проксимальных отделов ног. Отмечаются затруднения ходьбы по лестнице, подъема из положения присев на корточки. Типичен симптом Говерса — если пациент сидел на полу, то для того, чтобы подняться, он использует собственное тело как опору.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. Н. «крыловидные лопатки»), талия становиться очень тонкой (т. Н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий — при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. Н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Запор. Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Диагностика
Миодистрофия Эрба-Рота диагностируется неврологом и генетиком при сопоставлении данных анамнеза (возраст дебюта заболевания, последовательность развития симптомов), неврологического статуса пациента, ЭФИ нервно-мышечной системы, генеалогических данных, результатов анализа ДНК и микроскопического исследования мышечной ткани.
Дифференциальная диагностика
Дифференцировать миодистрофию Эрба-Рота приходится от других форм этого заболевания (прогрессирующей дистрофии Дюшенна, миодистрофии Дрейфуса и Беккера), дерматомиозита, полимиозита, бокового амиотрофического склероза, токсической миопатии и тд.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию — консультация пульмонолога.
Лечение
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и тд Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
Источник
Прогрессирующая мышечная дистрофия Эрба-Рота является одной из наследственных форм поражения мышечной ткани.
Начало заболевания обычно в возрасте 10-20 лет (возможен дебют до 40 лет); проходит около 10-15 лет до полного обездвиживания. Передача признака – по рецессивному типу, сцепленному с полом.
Информация для врачей. Мышечная дистрофия Эрба-Рота шифруется по МКБ 10 под кодом G71.0. При этом в диагнозе обязательно указывается стадия заболевания (1 – умеренные нарушения движений, 2 – затруднения возникают при ходьбе, выполнении легкой физической работе, 3 – параличи, контрактуры и т.д.). После этого указывается выраженность сопутствующих проявлений (снижение интеллекта, осложнений со стороны сердца), темп прогрессирования (быстрое, среднее или медленное). При утрате способности к передвижению обязательно указывается этот факт.
Причины
Единой точки рения на возникновение мышечных атрофий не существует. Доминирует наследственная теория. Точные биохимические механизмы патогенеза до конца не раскрыты.
Симптомы
 Симптомы заболевания вначале не специфичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Симптомы заболевания вначале не специфичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Достаточно рано изменяется походка. Она становится похожей на «утиную» — переваливание ног из-за слабости мышц бедра и тазового пояса. Быстро развивается гипотрофия мышц верхнего плечевого пояса. Также постепенно развивается общая гипотрофия, а затем и атрофия мышц. Иногда имеет псевдогипертрофия голеней – замена мышечной массы жировой и соединительной тканью.
Со временем пациент перестает выполнять многие активные движения. Значимо затрудняется вставание, больным приходится вставать на четвереньки, помогать руками при вставании. Лицо пациента становится амимично, неполностью смыкаются веки, губы же, напротив, выворачиваются кпереди и нередко утолщаются (губы тапира). Мимику такого пациента ещё иногда называют лицом миопата.
Диагностика
Диагноз заболевания обычно выставляется достаточно точно. При постановке диагноза дистрофия Эрба-Рота обращают внимание на возраст в дебюте заболевания, наследственность, скорость прогрессии процесса. В неврологическом осмотре выявляется снижение рефлексов вплоть до выпадения, снижение тонуса мышц, наличие контрактур суставов (из-за неравномерного процесса атрофии мышц).
Вопреки заблуждениям, фасцикулярных подергиваний мышц не бывает. При регистрации биотоков мышц снижается амплитуда, но не частота разрядов. По ЭНМГ определяется укорочение длительности потенциалов действия, полифазность записи.
Биохимически нередко обнаруживается изменение активности креатиникиназы, АСТ и других ферментов. Иногда обнаруживается изменение состава электролитов крови.
Достоверным считают диагноз при проведении гистологического исследования мышц. Меняются форма и размеры мышечных волокон, изменяется восприятие их окраски гистологическими красителями, мышцы перерождаются, увеличивается объем мышечных ядер. Между мышечными волокнами определяется жир, соединительная ткань. При этом отсутствует пучковость распределение волокон, характерная для нейрогенных миопатий.
Лечение
Патогенетической терапии не разработана. Лечение симптоматическое и направлено на уменьшение скорости прогрессирования. Активно используют витамины группы В, витамин Е, АТФ, экстракт алоэ внутримышечно, АТФ. Некоторое время назад использовался анаболический гормон ретаболил, однако часто отмечалось усиление распада мышечной ткани. Также применяют такие препараты, как тиоктовая кислота, рибоксин, актовегин.
Важная роль отводится немедикаментозным методикам воздействия. Массаж пациентам с дистрофией Эрба-Рота должен проводиться в легком темпе, направлен на борьбу с мышечным спазмом, укрепление мышц. Также важная роль принадлежит ЛФК. ЛФК при заболевании должна быть умеренной, но регулярной, в идеале ежедневной. Тренируются все группы мышц.
Постоянство проведение профилактических мероприятий позволяет длительно сохранять возможность больных к самообслуживанию. В моей практике вспоминается пациентка, которая при дебюте заболевания в 25 лет до 60 сохранила способность к самостоятельному передвижению, самообслуживанию, пускай с ограничением.
Прогноз
Прогноз при всех мышечных дистрофиях, как правило, неблагоприятный. Заболевание постепенно прогрессирует, охватывая все группы мышц. Рано или поздно наступает обездвиживание пациента. Хотя при этом само заболевание практически не приводит к смерти пациента. Смерть обусловлена пролежнями, инфекциями легких, мочевыводящих путей и т.п.
Источник
Быстрая постановка диагноза и адекватное лечение позволяет пациентам с миопатией Эрба-Рота избежать глубокой инвалидности, смертельных осложнений. Патология редкая, причины кроются в генетических нарушениях, симптомы сходны со многими нервно-мышечными болезнями. Поэтому нельзя заниматься самолечением. При первых признаках надо сразу обследоваться у невролога и проконсультироваться с независимыми медицинскими экспертами.
О заболевании
Конечностно-поясной миодистрофией или миопатией называют прогрессирующую болезнь с поражением мускульных клеток (миоцитов). В процессе развития меняется структура мышц из-за метаболических нарушений, дисбаланса аминокислот и ферментов. Причиной является влияние мутированного гена на синтез белков, из которых образуются миоциты.
В название миодистрофии включили имена невропатологов Вильгельма Генриха Эрба и Владимира Карловича Рота. Эти врачи независимо друг от друга занимались изучением мышечной патологии.
Общие характеристики Эрба-Рота миодистрофии:
- впервые болезнь проявляется в подростковом возрасте;
- встречаются ювенильные формы заболевания, с дебютом до 7 лет;
- реже миопатия развивается после 20 лет;
- вначале отмечают поражение (ослабление) мускул плечевого либо тазового пояса;
- прогрессирование приводит к ухудшению функций и истончению вовлеченных в патологию мышц;
- в патологический процесс вовлекаются мышцы торса;
- на фоне миопатии возникает дыхательная, сердечная недостаточность, учащаются пневмонии;
- инвалидность диагностируют не раньше 15 лет с момента дебюта миопатии;
- умственная деятельность и интеллект не страдают;
- встречается не более 3 случаев на 100000 человек;
- патологии одинаково подвержены мужчины и женщины;
- чем раньше поставят диагноз и начнут лечение, тем дольше человек будет способен двигаться;
- в зоне риска находятся люди до 30 лет, с аномалиями генов, с миопатиями у родственников.
Мышечной дистрофии Эрба-Рота характерно наследование по аутосомно-рецессивному типу. То есть, родители здоровы, но они – носители мутированного неполового гена. При совокупности факторов миопатия может развиваться у их одного ребенка или всех детей. У внуков же болезнь не проявится (это носители аномального гена), а у правнука(ов) будет патология.
Справка! Согласно международной классификации болезней код патологии по МКБ-10: G71.0.
Миопатия Эрба-Рота у ребенка прогрессирует, только если оба родителя носят мутированный ген. В случае, когда мать или отец здоров, болезнь может не проявиться, у внуков вероятность миопатии равна 25%.
Синонимичные названия болезни Эрба-Рота: юношеская миопатия, ювенальная мышечная атрофия, ПКМД. Патологию в МКБ-10 обобщили с другими первичными миодистрофиями под кодом «G71.0». После цифры врач уточняет стадию, степень нарушений, скорость прогрессирования, осложнения.
Причины развития
Этиологию и механизм развития миодистрофии продолжают изучать. У всех пациентов с миопатией Эрба-Рота генетическим исследованием выявлено мутацию неполовых генов. В большинстве случаев такая аномалия и высокая частотность зафиксирована в семьях, где был инцест — рождение детей от кровных родственников.
В основе патогенеза развития миопатии Эрба-Рота – изменения в генных локусах — местах хромосом:
- 5q33;
- 15q15―q21.1;
- 13q12;
- 17q12-q21.33.
В 30% случаев генная мутация не связана с наследственностью. Возможно, аномалию провоцирует радиация, плохая экология (отравление токсическими веществами) и подобные факторы.
Симптомы
Нет строгого правила развития и течения болезни. Миодистрофия может дебютировать в любое время между рождением и 40 годами. Вначале изменения могут отмечаться в тазовом либо плечевом поясе. В первом случае схема распространения называется восходящей, во втором – нисходящей.
Симптомы проявления миодистрофии Эрба-Рота в мышцах поясницы, таза:
- человек передвигается вперевалку (утиная походка);
- нарушается способность удерживать равновесие;
- ослабляется мускулатура ног;
- тяжело подниматься из позы «на корточках» или со стула;
- трудно ходить по неровной поверхности, лестнице;
- при вставании с пола пациент выпрямляется, опираясь руками поочередно на голени, колени, бедра (симптом Говерса);
- по мере прогрессирования сначала поражаются мышцы туловища, затем вовлекаются плечи, руки;
- угасает подошвенный, коленный рефлекс.
 Явные симптомы миопатии Эрба-Рота
Явные симптомы миопатии Эрба-Рота
При нисходящем типе миодистрофии Эрба-Рота мышечную гипотонию отмечают в плечах, верхних конечностях. Тяжело поднимать руки верх, расчесываться, мыть голову, надевать шапку. По мере распространения поочередно вовлекается мускулатура рта, век, грудной клетки, пояса, таза. Потом ослабевают ноги.
Симптомы прогрессирующей мышеной дистрофии Эрба-Рота с поражением плечевого пояса:
- невозможность удержать предмет, поднять ко рту ложку, завести руки за голову;
- свободные надплечья (плечи легко смещаются к уровню ушей, если человека приподнимают за подмышки);
- крыловидные лопатки (выпирают из-под кожи);
- лицо сфинкса (прикрытые веки, утолщенные губы);
- выпячивание живота вперед;
- осиная талия (неестественно тонкий обхват туловища на уровне пояса);
- искривление хребта в пояснице (гиперлордоз);
- ухудшается рефлекс сухожилия трицепса, бицепса.
Прогрессирование миопатии бывает быстрое, со средним и медленным темпом изменения мускульных волокон. При миодистрофии Эрба-Рота вначале слабеют пораженные мышцы. По мере развития теряются их функции, уменьшается объем (атрофируются, истончаются). Эта болезнь оканчивается инвалидностью или ограничением движения, всегда сопровождается повышенной физической усталостью.
Справка! Рекомендуется обоим родителям пройти тест на гетерозиготное носительство. Генное исследование ДНК поможет оценить риск рождения детей с миопатией.
Диагностика
На обследование идут к неврологу или ревматологу. В ходе осмотра врач проверяет функционирование нервной системы, рефлексы, чувствительность кожи, телосложение пациента. Для определения состояния мышц проводят электронейромиографию. При подозрении Эрба-Рота миодистрофии выполняют биопсию мускульных волокон для цитоморфологии. Нужен общий и биохимический анализ крови, исследование концентрации креатинфосфокиназы, состава мочи.
Доктор также даст направление к генетику для выявления генных мутаций, провоцирующих миодистрофию Эрба-Рота.
Обязательно проводят дифференциальную диагностику с другими мышечными дистрофиями – клиническое течение Эрба-Рота и Дюшена иногда может протекать практически одинаково.
Для исключения возможных осложнений болезни делают флюорографию, электрокардиограмму, УЗИ мышц, сердца, органов брюшной полости и малого таза, рентгенографию грудной клетки.
Диагностические признаки Эрба-Рота миодистрофии:
- мышцах рук/ног отмечается гипотония (ослабление тонуса);
- уменьшается объем пораженной мускулатуры;
- ухудшаются или исчезают в конечностях сухожильные рефлексы;
- по нервным волокнам продолжают передаваться импульсы;
- нейроны не повреждаются;
- в мышцах выявляют разной толщины мускульные волокна, соединительную ткань, отмирание миоцитов;
- в мышечной ткани дефицит креатинфосфокиназы;
- возможно изменение электролитного состава крови;
- через время после дебюта в крови нормализуется показатель креатинфосфокиназы.
На осложнения миопатии указывает воспаление легких, дистрофия миокарда, истончение гладких мышц внутренних органов. На ЭКГ выявляют ухудшение проводимости, изменение ритмичности сердца.
Лечение
Болезнь неизлечима, поскольку только изучаются способы устранения генной аномалии. Терапевтические методы направлены на торможение прогресса миодистрофии Эрба-Рота, купирование симптомов, улучшения метаболизма. Упор делают на рациональное питание, прием витаминно-минеральных комплексов, массаж, ЛФК, физиотерапию.
Что назначают при Эрба-Рота миодистрофии:
- витамины E, группу B, D, C, A;
- аденозинтрифосфорную кислоту;
- внутримышечные инъекции экстракта алоэ;
- метаболики — Актовегин, Рибоксин, Тиоктовая кислота;
- препараты галантамина;
- антиоксиданты, антигипоксанты — средства триметазидина;
- введение стволовых фетальных клеток;
- хлорид кальция — для ионофореза;
- Прозерин — для электрофореза;
- антидепрессанты 4 поколения с эсциталопрамом, флуоксетином.
Справка! При болезни Эрба-Рота прекратили использовать «Ретаболил» и другие гормональные препараты нандролона. В ходе клинических наблюдений выявлено ускорение распада мускульных волокон.
При Эрба-Рота миодистрофии пациент должен ежедневно употреблять белковую пищу (мясо, рыбу) и свежие фрукты, овощи, питательные смеси с аминокислотами, L-карнитином. Их вещества используются организмом для построения мускульных волокон. Важно пить минеральную воду (разновидность и суточную норму врачи подбирают персонально). Разрешено после консультации с доктором применять народные средства.
Для улучшения состояния здоровья полезны:
- чай лапачо — кора муравьиного дерева;
- настой плодов шиповника;
- отвар девясила;
- мед;
- цветочная пыльца.
При Эрба-Рота миодистрофии делают ежедневно упражнения ЛФК, дыхательную гимнастику и массаж. Плавают не реже 1 раза/неделю. Методы используют для укрепления и увеличения объема мышц, нормализации питания клеток, микроциркуляции. Это улучшает метаболизм, сохраняет способность двигаться, убирает застой жидкости в органах, мягких тканях.
Принципы ЛФК при Эрба-Рота миодистрофии:
- нагрузку и упражнения подбирает врач согласно состоянию здоровья;
- темп выполнения и интенсивность умеренные или средние;
- тренируют все группы мышц;
- людям с ограничениями упражнения помогают делать близкие;
- длительность тренировки не должна превышать 20 минут;
- большой комплекс упражнений разбивают на 2―3 раза/день.
Поддерживающий массаж выполняют с легким нажимом минимум полчаса. Используют разминающие, поглаживающие, растирающие, точечные, продольные движения. При уходе за инвалидом надо чаще, интенсивнее массировать зону грудной клетки, чтобы предупредить развитие застойной пневмонии.
Для растяжения укороченных мышц делают элонгирующий массаж. При Эрба-Рота миодистрофии такой сеанс проводят от 20 минут, нажимают сильнее, используют растягивающие, разминающие, вибрирующие движения.
Осложнения лечат этиологическими лекарствами и методами в зависимости от того, какой орган затронут. Например, в случае пневмонии используют антибиотики, при дыхательной недостаточности назначают искусственную вентиляцию легких.
Прогноз и осложнения
Нет точно установленного течения мышечной дистрофии. У одних миопатия быстро прогрессирует, вызывает скорую инвалидизацию. Другие с болезнью Эрба-Рота живут несколько десятилетий, могут самостоятельно себя обслуживать, но есть физические ограничения.
Тяжелое течение и быстрое прогрессирование болезни существенно сокращает продолжительность жизни пациентов. Смерть чаще фиксируют из-за осложнений миопатии. При Эрба-Рота болезни возможна недостаточность или дистрофия миокарда, нарушение газообмена в легких, аритмия, застойная пневмония, другие сердечные или дыхательные расстройства.
Вовлечение гладкой мускулатуры ниже диафрагмы ухудшает моторику желудка, перистальтику кишечника, функции мочевого пузыря, тонус матки. Развиваются болезни мочеполовой системы, появляется недержание мочи, хронический запор, другие нарушения работы пищеварительных и тазовых органов.
Профилактика
К первичной профилактике относят отказ от брака с кровным родственником, соблюдение правил безопасности при работе с вредными веществами. Людям с подтвержденным гетерозиготным носительством следует помнить о риске рождения детей с миодистрофией. Их ребенка врачи должны наблюдать с младенчества, а родителям надо ежедневно делать малышу гимнастику и массаж.
Важно! ЛФК, лечебное питание, массаж и методы физиотерапии помогают затормозить прогресс миодистрофии Эрба-Рота в любом возрасте. Проводится только профилактика осложнений.
Что нужно запомнить?
- Миопатия относится к первичным мышечным болезням.
- Причиной является генная мутация.
- Симптомы разнообразны, но Эрба-Рота миодистрофия всегда затрагивает мускулатуру конечностей, плеч, поясницы.
- В ходе обследования изучают функции и структуру мышечной ткани.
- Лечат препаратами симптоматической терапии, немедикаментозными методами.
- Прогноз качества жизни неблагоприятный, смерть наступает из-за осложнений.
- Не разработана специфическая профилактика миодистрофии Эрба-Рота.
Литература
- Гланц С. Медико-биологическая статистика. // М. Практика. 1999. 459 с.
- Зенгбуш П. Молекулярная и клеточная биология. // Из-во «Мир». М. 1982. т.1,2,3.
- Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. // М. Из-во «Мир». 1993, том 1 и 2.
- Шишкин С.С. Наследственные нервно-мышечные болезни. // Изд-во ВИНИТИ, Москва, 1997, 130с.
- Шубникова Е.А. Строение, развитие, сравнительная и патологическая гистология мышечных тканей. // В кн.: «Мышечные ткани» под. ред. Ю.С. Ченцова Из-во «Медицина». М. 2001. 7-108.
- Berg J.S., Powell B.C., Cheney R.E. A millenial myosin census. // Mol. Biol. Cell. 2001. V.12. P.780-794.
- Emery A.E.H. Diagnostic Criteria for Neuromuscular Disorders. // European Neuromuscular Centre, The Netherlands, 1994. 72p.
- Kaplan J.C., Fontaine B. Neuromuscular disorders: gene location. // Neu-romusc. Disord. 1999. V.9. P.I-VIII.
- Redowicz M.J. Myosins and pathology: genetics and biology. // Acta Bio-chim. Polonica 2002 V.49. P.789-804.
Источник