Миопатический синдром код по мкб

Содержание
- Описание
- Дополнительные факты
- Симптомы
- Патогенез
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Миопатии.
Миопатии
Описание
Миопатии. Группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Дополнительные факты
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Особенности отдельных форм миопатии.
Ювенильная миопатия Эрба наследуется аутосомно. Рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече. Лопаточно — лицевая миопатия Ландузи — Дежерина имеет аутосомно — доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия. Аутосомно — доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно. Доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
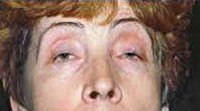
Миопатии
Симптомы
Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Патогенез
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр. ), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и тд ), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гипотиреоза, гипертиреоза, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и тд формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные — возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при синдроме Шегрена, СКВ, склеродермии и тд коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов — уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и тд ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
- Описание
- Причины
- Симптомы (признаки)
- Лечение
Краткое описание
Миопатии — обобщённое название множества заболеваний мышечной системы (главным образом, скелетной мускулатуры), обусловленных нарушением сократительной способности мышечных волокон и проявляющихся мышечной слабостью, уменьшением объёма активных движений, снижением тонуса, атрофией, иногда псевдогипертрофией. Точная диагностика миопатий сложна, во многих случаях необходима биопсия мышц, практически во всех случаях обязателен углублённый семейный анамнез. Классификация миопатий запутана, многие нозологические единицы имеют эпонимические, слишком общие и частично совпадающие наименования. Обычно выделяют первичные (наследственные) и вторичные миопатии. Первые включают в себя врождённые структурные миопатии (см. ниже) и прогрессирующие мышечные дистрофии (см. Дистрофии мышечные, Дистрофия мышечная Дюшенна). Вторые объединяют поражения мышц при различные системных заболеваниях (коллагенозы, эндокринные нарушения и пр., см. также Миопатии метаболические).
Код по международной классификации болезней МКБ-10:
- G71.2 Врожденные миопатии
- G71.3 Митохондриальная миопатия, не классифицированная в других рубриках
- G72 Другие миопатии
Причины
Генетические аспекты. Типы, гены и дефект: • недостаточность карнитин пальмитоилтрансферазы II, 255110, CPT2, 600650, 1p32; • недостаточность сукцинат дегидрогеназы, SDH1, 185470, 1p22.1 qter; • недостаточность фосфоглицерат мутазы, PGAM2, PGAMM, 261670, 7p13 p12.3; • Броди, 601003, ATP2A1, SERCA1, 108730, 16p12; • врождённая доброкачественная, 158810, ген коллагена COL6A1, 120220, 21q22.3; • врождённая доброкачественная, 158810, ген коллагена COL6A3, 120250, 2q37; • дистальная, MPD1, 160500, 14q; • миотубулярная (центронуклеарная) X сцепленная, MTM1, MTMX, 310400, Xq28; • Миоши, 254130, LGMD2B, 253601 (тазово плечевая дистрофия мышечная), 2p13.3 p13.1; • немалиновая, тип 1, 161800, NEM1, 191030 (тропомиозин 3, TPM3), 1q22 q23; • немалиновая, тип 2, 256030, NEM2, 191030 (тропомиозин 3, TPM3), 1q22 q23; • с тельцами включения (), IBM2, 601073, 9p1 q1; • 125660, ген десмина DES, 2q35.
Некоторые формы врождённых миопатий
• Немалиновая — непрогрессирующая мышечная слабость, чаще поражающая проксимальные мышцы. В мышечных волокнах характерные палочковидные и нитеобразные стержни (элементы Z — линий). Синонимы: миопатия врождённая непрогрессирующая, миопатия нитеобразная.
• Центронуклеарная (миотубулярная) — медленно прогрессирующие мышечная слабость и атрофия, начинающиеся в детском возрасте; ядра большинства мышечных волокон локализованы преимущественно в центральной части, нежели по периферии мышечных волокон.
• Болезнь центрального стержня — медленно прогрессирующая слабость мышц; при биопсии в сердцевине мышечных волокон отсутствуют митохондрии и элементы саркоплазматической сети, активность окислительных ферментов, фосфорилазы, АТФазы; миофибриллы расположены в виде компактных образований.
• Митохондриальные — разнообразная группа редко встречающихся заболеваний, обусловленных дефектами митохондриального и/или ядерного генома. На первый план выступают прогрессирующие мышечная слабость и утомляемость, часты миокардиодистрофии, симптоматика со стороны ЦНС (задержка психомоторного развития, деменция, интермиттирующая кома, судорожные припадки), а также симптомы поражения различных органов и систем. Диагностика затруднена, обязательно включает биопсию мышц.
• Митохондриальные энцефаломиопатии — разнородная группа заболеваний с общим морфологическим признаком — нарушением структуры митохондрий в мышцах и ЦНС, но различными молекулярными дефектами в виде нарушений транспорта субстратов из цитозоля в митохондрии, утилизации субстратов, структуры ферментов цикла Кребса, сопряжения окисления и фосфорилирования.
• Миопатия Броди (#601003, 16p12, ген ATP2A1 [SERCA1, Ca2+ — АТФаза саркоплазматического ретикулума быстрых фазных мышечных волокон], 108730, обычно ). Безболезненные спазмы мышц, нарушение расслабления мышц.
Симптомы (признаки)
Клиническая картина • Сходная для всех врождённых миопатий • Дети рождаются с низким мышечным тонусом и слабостью лицевой мускулатуры • Заболевание обычно не прогрессирует или регрессируют с возрастом • Иногда развивается дыхательная недостаточность • В подростковом периоде, как правило, развивается сколиоз. Диагноз устанавливают по результатам биопсии мышц.
Лечение
Лечение • В первые месяцы жизни ребёнка могут потребоваться борьба с дыхательными расстройствами и питание через зонд • Позже возникает необходимость в ортопедической коррекции, лечебной гимнастике, физиотерапии и рациональном трудоустройстве.
МКБ-10 • G71.2 Врождённые миопатии • G71.3 Митохондриальная миопатия, не классифицированная в других рубриках • G72 Другие миопатии
OMIM • 160560, 252010 Митохондриальная миопатия с гигантскими митохондриями • 251945 Митохондриальная миопатия с дефектами транспорта белка в митохондриях • 251950 Митохондриальная миопатия и молочнокислый ацидоз • 160550 Митохондриальная миопатия с ранней катарактой • 161800, 256030 Миопатия немалиновая • 191030 Миопатия немалиновая • 160500, 160300, 254130 Миопатия поздняя дистальная • 160150, 255200, 310400 Миопатия центронуклеарная • 117000 Болезнь центрального стержня • 160200 Миопатия врождённая с внутриядерными включениями кристаллина E • 160570 Миопатия с отложением гликопротеинов и гликозаминогликанов • 254940 Синдром Карей–Файнман–Зитера • 254950 Миопатия грануловакуолярная дольковая с миотонией • 254960 Миопатия вследствие дефекта малат — аспартатного шунта • 255100 Миопатия с нарушением метаболизма липидов • 255160 Миопатия с лизисом миофибрилл типа I • 255300 Миопатия врождённая Бюттена–Тёрнера • 255310 Миопатия врождённая с диспропорцией волокон • 255320 Миопатия врождённая многоцентровая с наружной офтальмоплегией.
Источник
Содержание
- Описание
- Дополнительные факты
- Классификация
- Причины
- Симптомы
- Диагностика
- Лечение
Названия
Название: Немалиновая миопатия.
Немалиновая миопатия
Описание
Немалиновая миопатия. Группа генетически разнородных наследственных миопатий, общим патогистологическим проявлением которых является образование в мышечной ткани нитевидных структур, что нашло свое отражение в названии патологии (от греческого Nema — нить). Симптомы различных форм состояния могут значительно различаться по своей выраженности – возможны гипотония мышц, мышечная слабость (в том числе дыхательных и мимических мышц), аномалии развития скелета (сколиозы, удлиненная форма черепа, расщелины твердого нёба). Диагностика основывается на оценке статуса больного, изучении биоптата мышечной ткани, молекулярно-генетических исследованиях. Специфического лечения не существует, используется поддерживающая и симптоматическая терапия.
Дополнительные факты
Немалиновые миопатии (NEM) – совокупность генетических патологий со сходными клиническими проявлениями, которые характеризуются выраженным нарушением структуры мышечной ткани с формированием в ней аномальных нитеподобных или палочковидных структур. Впервые заболевание было описано в 1963-м году двумя независимыми группами исследователей под началом Д. Шая и П. Конена, в последующие годы было выявлено множество генетических разновидностей данного состояния. В зависимости от гена и мутации в нем, немалиновая миопатия может иметь как аутосомно-доминантный, так и аутосомно-рецессивный характер наследования. Встречаемость патологии на сегодняшний день не определена, но, по мнению многих врачей-генетиков, она является одной из наиболее распространенных врожденных миопатий – иногда указываются цифры 1:50 000. При этом достоверно определить этиологию немалиновой миопатии (ответственный за ее развитие ген и характер его дефекта) удается только в половине случаев, что указывает на незавершенность изучения данного заболевания.

Немалиновая миопатия
Классификация
Причиной развития немалиновой миопатии являются генетические дефекты, приводящие к формированию аномальных форм структурных белков мышечной ткани – в основном, саркомерных протеинов. В результате этого нарушаются процессы сокращения мышц, а также, в ряде случаев, и их формирование, что приводит к появлению врожденных форм данного состояния. На сегодняшний день удалось идентифицировать семь генов, мутации которых ответственны за развитие немалиновой миопатии, однако, как было указано выше, они обуславливают лишь половину всех случаев заболевания. Кроме того, существует форма патологии с отсроченным началом, поражающая в основном взрослых людей – но, как показали более подробные исследования, она обусловлена приобретенными аутоиммунными нарушениями, а не генетическими факторами.
Помимо классификации, созданной на основе данных современной генетики, немалиновая миопатия также разделяется по своим фенотипическим проявлениям на врожденную (типичную, промежуточную и тяжелую разновидности) и ювенильную формы. Взаимосвязь этих двух систем классификации достаточно условна – различные мутации одного и того же гена могут приводить к развитию тяжелых и типичных врожденных форм или ювенильной разновидности заболевания.
NEM. 1 — форма немалиновой миопатии, обусловленная мутацией гена TPM3, расположенного на 1 — й хромосоме, регистрируется примерно в 2 — 3% случаев патологии. Ген кодирует одну из цепей медленного альфа-тропомиозина – структурного белка мышечной ткани. К немалиновой миопатии могут приводить различные мутации данного гена с разным типом наследования. Аутосомно-доминантные дефекты становятся причиной развития ювенильной формы заболевания, а аутосомно-рецессивные характеризуются врожденным тяжелым течением патологии.
NEM. 2 — наиболее распространенный вариант немалиновой миопатии, составляющий примерно половину от всех генетически идентифицированных случаев данного заболевания. Вызывается дефектом гена NEB, который локализован на 2-й хромосоме и кодирует гигантский белок небулин. Последний функционально связан с тонкими (актиновыми) нитями мышечного волокна. Мутации гена NEB приводят к немалиновой миопатии исключительно врожденного характера с типичным или тяжелым течением.
Причины
NEM. 3 — вторая по распространенности форма немалиновой миопатии, регистрируемая в 20 — 25% случаев заболевания с определенной этиологией. Причиной данной формы становятся мутации гена ACTA1, расположенного на 1-й хромосоме. Продуктом его экспрессии является альфа-актин скелетных мышц, один из важнейших структурных компонентов мышечной ткани. Выявлено более 180 типов мутаций данного гена, приводящих к самым разнообразным формам немалиновой миопатии с разным характером наследования, в основном – с врожденными нарушениями. Часть генетических дефектов ACTA1 являются спонтанными мутациями, возникающими de novo, по этой причине иногда возможна зародышевая мозаичность.
NEM. 4 — разновидность немалиновой миопатии, составляющая примерно 3 — 4% всех случаев данного заболевания. Обусловлена дефектом гена TPM2, локализованного на 9-й хромосоме и кодирующего белок бета-тропомиозин, входящий в группу саркомерных протеинов. Мутации этого гена чаще всего передаются по аутосомно-доминантному механизму, имеются указания на возможность спонтанного повреждения гена. Этот генетический дефект проявляется врожденной типичной разновидностью немалиновой миопатии.
NEM. 5 — форма немалиновой миопатии, диагностируемая исключительно в закрытых религиозных общинах амиш, где встречаемость этого заболевания достигает 1 — 500. Вызывается мутацией гена TNNT1, который располагается на 19-й хромосоме и отвечает за белок под названием медленный тропонин Т. Такие генетические нарушения приводят к тяжелой врожденной форме немалиновой миопатии с аутосомно-рецессивным механизмом наследования.
NEM. 6 — редкая и малоизученная разновидность немалиновой миопатии, которая предположительно вызывается мутацией гена KBTBD13, локализованного на 15 — й хромосоме. Достоверно неизвестно, какой именно белок кодирует данный ген, поэтому его роль в этиологии данного заболевания ставится под сомнение некоторыми исследователями. Предполагают, что его дефекты являются причиной аутосомно-доминантной немалиновой миопатии с легким течением и развитием в детском возрасте (ювенильная разновидность).
NEM. 7 — также редкая форма немалиновой миопатии, которая была описана лишь у одной семьи в 2007 — м году. Обусловлена дефектом гена CFL2, расположенного на 14-й хромосоме, продуктом его экспрессии является белок кофилин-2 (мышечный кофилин), принимающий активное участие в поддержании стабильного состояния актиновых нитей. Мутации CFL2 приводят к врожденной типичной форме заболевания с аутосомно-рецессивным характером наследования.
Симптомы
По своим клиническим проявлениям все случаи немалиновой миопатии разделяются на две группы – врожденную (которая, в свою очередь, делится на типичную, промежуточную и тяжелую) и ювенильную, для которой характерно появление первых симптомов в детском (8-13 лет) возрасте. Врожденная разновидность встречается намного чаще, может иметь как аутосомно-доминантный, так и рецессивный механизм наследования, тогда как ювенильная практически всегда бывает только аутосомно-доминантной. Больные немалиновой миопатией врожденного типа имеют характерный внешний вид – вытянутое лицо с бедной мимикой, приоткрытый рот, удлиненный череп.
Типичная (умеренная) разновидность является наиболее распространенной, сопровождается миотонией, затруднением при кормлении, антигравитационными движениями. Нарушения дыхания при этой форме немалиновой миопатии выражены незначительно, обычно сводятся к пониженной вентиляции легких по ночам. Слабость скелетных мышц в основном имеет проксимальный характер, прогрессирует крайне медленно, в ряде случаев больные способны передвигаться на костылях или ходить при помощи окружающих.
Промежуточная разновидность врожденной немалиновой миопатии представляет собой более тяжелую форму заболевания. Она характеризуется наличием всех вышеперечисленных нарушений, которые при этом имеют тенденцию к прогрессированию, возможны околосуставные ретракции, искривления позвоночника. Обычно к 6-7 годам больные этой формой немалиновой миопатии полностью утрачивают возможность передвигаться без инвалидного кресла, а к 10-12 годам – самостоятельно дышать, что приводит к летальному исходу. Тяжелая форма немалиновой миопатии проявляется резко выраженной миопатией в неонатальном периоде, затруднением глотания и сосания, в ряде случаев сильно страдает дыхательная мускулатура. Летальный исход наступает в первые месяцы жизни из-за дыхательной недостаточности или респираторной инфекции.
Диагностика
Основными методами диагностики немалиновой миопатии являются изучение наследственного анамнеза пациента, гистологическое исследование мышечной ткани, электромиография и молекулярно-генетические исследования. В анамнезе больного могут определяться случаи заболевания у родственников, отсутствие таких примеров указывает на спорадический характер мутации. Патогистологические изменения в мышечной ткани сводятся к наличию в саркоплазме волокон нитевидных включений – при этом они могут иногда не определяться в мышцах младенцев даже при врожденных формах заболевания и появляться в более старшем возрасте. Иногда при немалиновой миопатии может обнаруживаться также неоднородность толщины мышечных волокон и их частичное разрушение. Электромиография при этом заболевании имеет признаки характерной «миопатической триады» – уменьшение амплитуды и длительности потенциалов действия в сочетании с их полифазностью. При помощи достижений современной генетики можно определить некоторые формы немалиновой миопатии – обусловленные мутациями генов NEB, ACTA1 и TPM2.
Лечение
Специфического лечения немалиновой миопатии не существует, используют поддерживающую терапию, лечебную физкультуру, гимнастику для сохранения хотя бы минимальной активности мышц и предотвращения суставных контрактур. При нарушении дыхания применяют принудительную вентиляцию легких, питание младенцев в случае нарушений глотания или сосания осуществляют через назогастральный зонд. Если искривления позвоночника или другие скелетные нарушения могут усугубить состояние больного, прибегают к помощи хирургов-ортопедов.
Источник