Миоклония код по мкб 10

Рубрика МКБ-10: G25.3
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G20-G26 Экстрапирамидные и другие двигательные нарушения / G25 Другие экстрапирамидные и двигательные нарушения
Определение и общие сведения[править]
Синдром опсоклонус-миоклонус
Синонимы: синдром Кинсбурна, паранеопластический опсоклонус-миоклонус, POMA синдром
Синдром опсоклонус-миоклонус является редким нейровоспалительным заболеванием паранеопластическго, инфекционного или идиопатического происхождения. Характеризуется опсоклонусом, миоклонусом, атаксией, расстройствами поведения и сна.
Ежегодная заболеваемость оценивается на уровне около 1/5000000.
Этиология и патогенез[править]
В большинстве случаев паранеопластической этиологии у детей, обнаруживается нейробластома. Инфекции запускающие синдром опсоклонус-миоклонус включают множество вирусных и бактериальных агентов, включая стрептококки, микоплазмы и ветряную оспу. Точный патогенез неизвестен, но предполагается наличие опосредованной аутоиммунной стволовой и/или мозжечковая дисфункции. Опсоклонус может отражать растормаживание фастигиального ядра мозжечка или неупорядоченного взаимодействия между универсальными паузовыми и залповыми нейронами. Однако, когнитивные и поведенческие элементы патологии, а также недавние нейровизуалиционные исследования, позволяюь предложить наличие более широкого неврологического процесса.
Клинические проявления[править]
Синдром опсоклонус-миоклонус обычно манифестирует в возрасте от 1 до 3 лет. Он характеризуется опсоклонусом (быстрые, разнонаправленные движения глаз), миоклоническими подергиваниями, атаксией, раздражительностью и нарушениями сна. Течение может быть монофазное или хроническое рецидивирующее. Синдром опсоклонус-миоклонус связан приблизительно в 50% детских случаев с развитием нейробластомы. Опухоль, как правило (но не всегда) низкой степени злокачественности с хорошим прогнозом. Во взрослом возрасте чаще всего обнаруживается мелкоклеточный рак легкого и аденокарциномы молочной железы.
Миоклонус: Диагностика[править]
Диагноз клинический, основанный на наличии 3 из 4 следующих критериев: 1) нейробластома, 2) опсоклонус, 3) двигательные расстройства с миоклонусом и/или атаксией, и 4) нарушениями поведения и/или сна.
Нейробластома выявляется при детальном МРТ с особым акцентом к параспинальной области, каротидному синуса, средостению, надпочечникам, полости живота и таза. Нейробластомы при патологии, как правило, метаболически неактивны. Серологические тесты могут помочь идентифицировать случаи параинфекционной этиологии. У взрослых обнаруживаются Hu анти-нейрональные ядерные антитела (анти-Hu).
Дифференциальный диагноз[править]
Дифференциальный диагноз включает острую воспалительную мозжечковую атаксию, которая по типу движения глаз (нистагм), отсутствию раздражительности, и, как правило, быстрым восстановлением без лечения.
Миоклонус: Лечение[править]
Лечение обычно включает в себя резекцию нейробластомы, иногда, может потребоваться химиотерапия. Лечение включает в себя иммунокоррекцию. Схемы лечения не были стандартизированы, но могут включать в себя кортикостероиды, адренокортикотропный гормон гормон, циклофосфамид, внутривенный иммуноглобулин, и/или ритуксимаб.
Прогноз
Некоторые дети хорошо реагируют на стероиды и не имеют осложнений. Другие могут быть резистентными к лечению, имеют хроническое рецидивирующее течение с двигательными, когнитивными и/или поведенческими нарушениями. Опсоклонус обычно ослабевает. Наличие или отсутствие нейробластомы, видимо, не влияет на прогноз.
Профилактика[править]
Прочее[править]
Эссенциальная семейная доброкачественная миоклония
Этиология и патогенез
Наследуется по аутосомно-доминантному типу, однако возможны и спорадические случаи. Парамиоклонии в 1881 г. описал Н. Фридрейх (Friedreich N., 1825-1882) под названием «множественная парамиоклония», известная в дальнейшем как парамиоклония Фридрейха. Миоклонии в отличие от парамиоклоний относятся к быстрым по темпу гиперкинезам и сопровождаются изменением частей тела в пространстве.
Нейрохимические исследования позволяют считать, что миоклонии и парамиоклонии связаны с поражением серотонинергических нейронов ядра шва или исходящих отсюда путей, направляющихся к промежуточному и конечному мозгу, а также к спинному мозгу. В результате из-под контроля тормозящих серотонинергических нейронов высвобождаются церебральные и спинальные структуры, обладающие облегчающим влиянием на моторную функцию. Предполагается, что такими структурами являются гигантоклеточное ядро продолговатого мозга, оказывающее облегчающее влияние на альфа-мотонейроны, и зоны ретикулярной формации ствола, участвующие в активации вышележащих структур мозга. Серотонинергические нейроны ядра шва оказывают тормозное влияние на проводимость импульсов по афферентным и эфферентным проводящим путям.
Клинические проявления
Эссенциальная (наследственная) доброкачественная миоклония характеризуется диффузными гиперкинезами по типу парамиоклонии (сокращений отдельных мышечных пучков) без изменения положения частей тела в пространстве. Гиперкинезы чаще проявляются в мимических мышцах и в мышцах конечностей. Миофасцикуляции ритмические (миоритмии) с частотой от 10 до 50 в минуту возникают периодически в разных мышцах, в большинстве случаев симметричны, хотя и не всегда синхронны. Сухожильные рефлексы обычно равномерно оживлены, чувствительность — без особенностей. Возникает, как правило, в зрелом возрасте; течение хроническое, доброкачественное.
Лечение
В лечении миоклоний и парамиоклоний нередко эффективно введение предшественников серотонина: L-5-гидроокситриптофан (от 400 до 2000 мг/сут), L-триптофан, окситриптофан, начиная от 125 мг/сут с последующим наращиванием дозы диазепама (до 15-20 мг/сут). Эффективность повышается при одновременном применении препаратов эльдопы. Побочные явления: анорексия, тошнота, диарея, особенно на начальном этапе лечения, возможны нарушения дыхания — гипервентиляция или диспноэ. В таких случаях необходимо снижение дозы предшественников серотонина и назначение антигистаминных средств.
Наряду с предшественниками триптофана целесообразны клоназепам. Подбор их дозы индивидуален. Для взрослых обычная доза от 2 до 6 мг/сут. Побочные явления: вялость, сонливость, анорексия, атаксия. При спинальных миоклониях есть положительный опыт применения триметина. Иногда эффективны антихолинергические средства. Кроме того, возможен значительный эффект при лечении тиапридом в дозе 100-600 мг/ сут, пирацетамолом.
Наследственный гениоспазм
Синонимы: врожденный тремор подбородка, семейное подергивание подбородка
Определение и общие сведения
Наследственные гениоспазм является двигательным расстройством, которое характеризуется эпизодами непроизвольного тремора подбородка и нижней губы.
Расстройство было описано в менее чем 25 семей из Европы и США, с небольшим преобладанием мужского пола (отношение мужчины к женщине 1,3: 1).
Наследование аутосомно-доминантное.
Этиология и патогенез
Локус был идентифицирован на 9q13-Q21 в одном четырех поколений британской семьи. Тем не менее, никакой связи с этим локусом не было обнаружено у другой британской семьи с заболеванием, что указывает на генетическую гетерогенность патологии.
Клинические проявления
Манифестация обычно происходит в детстве и может быть спровоцирована стрессом и эмоциями. Эпизоды могут возникнуть во время сна. Отсутствуют какие-либо сопутствующие неврологические нарушения, хотя аномальная ЭЭГ, расстройства сна и вовлечение других мышц лица были также описаны. Возможно спонтанное улучшение с возрастом.
Доброкачественный ночной миоклонус новорожденных
Синонимы: Benign neonatal sleep myoclonus
Доброкачественный ночной миоклонус новорожденных является неэпилептическим расстройством. Доброкачественный ночной миоклонус новорожденных возникает в первые недели жизни ребенка и характеризуется беспорядочными миоклоническими подергиваниями во время сна и не сопровождается изменениями на ЭЭГ.
Доброкачественный ночной миоклонус новорожденных не связан с осложнениями перинатального периода, разрешается самостоятельно в течение двух-четырех месцны и не несет угрозу развитию ребенка.
Источники (ссылки)[править]
https://www.orpha.net
Частная неврология [Электронный ресурс] / А. С. Никифоров, Е. И. Гусев. — 2-е изд., испр. и доп. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970426609.html
Неврология [Электронный ресурс] / Под ред. Е.И. Гусева, А.Н. Коновалова, А.Б. Гехт — М. : ГЭОТАР-Медиа, 2016. — https://www.rosmedlib.ru/book/ISBN9785970428900.html
Epileptic Disord. Volume 10, issue 2, June 2008
Дополнительная литература (рекомендуемая)[править]
1. Penn, R. D. Medical and surgical treatment of spasticity. Neurosurg. Clin. North Am. 1 (3):719, 1990.
Действующие вещества[править]
- Пирацетам
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Классификация
- Симптомы
- Возможные осложнения
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Профилактика
Названия
Название: Миоклоническая эпилепсия.
Миоклоническая эпилепсия
Описание
Миоклоническая эпилепсия. Заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.
Дополнительные факты
Миоклонические судороги (миоклонии) представляют собой непроизвольные сокращения отдельной мышцы/мышечной группы. Соответственно, эпилепсия с преобладанием в клинической картине миоклоний получила название миоклоническая. Понятие «миоклоническая эпилепсия» (МЭ) включает ряд заболеваний, разнородных по этиопатогенезу, возрасту дебюта, особенностям симптоматики. В подавляющем большинстве случаев они характеризуется сочетанием миоклоний и генерализованных тонико-клонических судорожных приступов, имеют генетическую обусловленность. Встречаемость МЭ различна, некоторые нозологические формы являются настолько редкими, что в литературе по неврологии описано не более 100 клинических случаев.
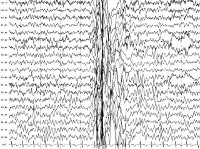
Миоклоническая эпилепсия
Причины
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
• Внутриутробные инфекции. Инфекционный процесс неблагоприятно отражается на развитии плода. Особенно опасны вирусные инфекции, поскольку вирусы способны провоцировать аномальную перестройку отдельных генов.
• Хронические заболевания беременной. Сахарный диабет, сердечная недостаточность, хронические заболевания лёгких, эндокринная патология матери приводят к гипоксии, метаболическим расстройствам на ранних стадиях развития зародыша. В результате происходят сбои формирования ЦНС, отдельных механизмов обмена веществ.
• Повышенный радиоактивный фон. Радиация оказывает мутагенное влияние на живые организмы. Развивающийся плод наиболее подвержен подобному воздействию. Следствием является возникновение структурных, дисметаболических, функциональных аномалий, влекущих за собой повышенную эпилептическую активность.
• Прием тератогенных медикаментов. Самолечение, незнание о своей беременности в раннем периоде, медицинская необходимость фармакотерапии приводят к приёму опасных для плода медикаментов. Химические вещества оказывают повреждающее воздействие на отдельные гены, вносят изменения в существующие метаболические механизмы.
• Токсические воздействия на плод. Алкоголизм, наркомания, курение женщины в период беременности сопровождаются проникновением токсических веществ в организм плода. Подобно тератогенным фармпрепаратам они способны повредить отдельный локус генома, в результате возникает миоклоническая эпилепсия.
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков). При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
• Идиопатические. Наследственно обусловленные формы. Характерна манифестация симптоматики в детском/подростковом возрасте. Идиопатическими являются доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ), юношеская миоклоническая эпилепсия (ЮМЭ), болезнь Унферрихта-Лундборга, синдром Драве.
• Криптогенные. Не имеющие установленной этиологии. Отличаются выраженной резистентностью к фармакотерапии, наличием сопутствующей очаговой симптоматики, интеллектуального дефицита. К криптогенным относятся эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами.
• Симптоматические. Возникающие на фоне происходящих в организме патологических процессов. В большинстве случаев обусловлены метаболическими нарушениями. Симптоматическими считаются ранняя миоклоническая энцефалопатия, болезнь Лафоры, миоклонические пароксизмы при подостром склерозирующем панэнцефалите, болезни Крейтцфельдта-Якоба.
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Возможные осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
Миоклоническая симптоматика входит в клинику многих болезней, эпилептических синдромов. Диагноз «миоклоническая эпилепсия» устанавливается только при превалировании миоклонических приступов над другими клиническими проявлениями. Диагностика направлена на верификацию нозологической формы эпилепсии, при выявлении вторичного характера миоклоний — на поиск основной патологии. Основными диагностическими этапами являются:
• Сбор анамнестических данных. Большое значение имеет возраст дебюта, характер начала, порядок развития симптоматики.
• Неврологический осмотр. Проводится неврологом, направлен на выявление миоклонических сокращений, очагового дефицита, определение уровня психического развития, степени когнитивных расстройств, оценку психического статуса.
• Электроэнцефалография. У большинства пациентов регистрируются диффузные интериктальные симметричные эпилептогенные разряды, иктальные высокоамплитудные спайки. В ряде случаев для выявления эпиактивности требуется суточный ЭЭГ-мониторинг, проведение провокационных проб (ЭЭГ при вспышках света, гипервентиляции, резких звуковых сигналах). Результаты исследований оцениваются нейрофизиологом, эпилептологом.
• Нейровизуализация. До закрытия родничков осуществляется путём нейросонографии, у детей старше года — при помощи МРТ головного мозга. Взрослым может проводиться МСКТ. Морфологические изменения церебральных тканей характерны для симптоматических МЭ.
• Лабораторные исследования. Производятся при подозрении на наличие обменных расстройств. Включают биохимический анализ крови и мочи, специфические анализы.
• Консультация генетика. Сбор семейного анамнеза, составление генеалогического древа позволяют определить наследственный характер эпилепсии, установить тип наследования.
Дифференциальная диагностика
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина.
Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
Профилактика
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Источник