Миелодиспластический синдром моносомия 7 хромосомы

Хромосомные аномалии при миелодиспластическом синдроме — прогноз
Кариотип клеток костного мозга у больных с миелодиспластическими синдромами (МДС) интенсивно изучается на протяжении последних 10—15 лет. Аномальные клоны выявлены до лечения у 30—50 % пациентов, в некоторых сообщениях приведены более высокие показатели — 60—75 %.
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны, спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
Наиболее характерны моносомии 5 и 7, а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20—30 %, при появлении начальных признаков трансформации в острый лейкоз — до 40—60 %, при трансформации в острый миелобластный лейкоз — 80—90 %.
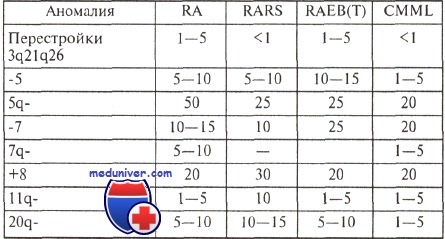
Транслокации, специфичные для первичных острых нелимфобластных лейкозах, при миелодисплазиях наблюдаются редко. Есть сообщения о повторяющихся транслокациях t(3;3)(q21;q26), t(8;21)(q22;q22) и t(3;21)(q26;q22). Примеры перестроек длинного плеча хромосомы 3 показаны на рисунке.
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Основные хромосомные аномалии, характерные для миелодисплазий:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q — рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге — признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974—1985 гг..
Делеции длинного плеча хромосомы 5 наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие — плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания, посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Другие аномалии определяли «промежуточный» прогноз. Средняя длительность заболевания до перехода в острый лейкоз составила по группам 9,4; 0,4 и 1,1—3,3 года соответственно. Эти данные используются при оценке эффективности новых схем лечения миелодисплазии и зарекомендовали себя как одна из лучших прогностических систем при миелодиспластическом синдроме.
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.
— Читать далее «Хромосомные аномалии при хроническом миелолейкозе (ХМЛ) — прогноз»
Оглавление темы «Цитогенетика лейкозов»:
- Транслокация (8;14)(q24;q32) при остром лимфобластном лейкозе
- Транслокация (1;19)(q23;p13) при остром лимфобластном лейкозе
- Транслокация (12;21)(p13;q22) при остром лимфобластном лейкозе
- Делеция 6q (6q-), 9р (9р-) при остром лимфобластном лейкозе
- Прогностическое значение хромосомных аномалий при остром лимфобластном лейкозе
- Хромосомные аномалии при миелодиспластическом синдроме — прогноз
- Хромосомные аномалии при хроническом миелолейкозе (ХМЛ) — прогноз
- Цитогенетика бластного криза хронического миелоидного лейкоза — изменения кариотипа
- Дифференциальная диагностика хронического миелолейкоза
- Молекулярно-генетические методы оценки эффективности лечения хронического миелолейкоза
Источник
Материалы представлены из учебного пособия РУДН
Анемии. Клиника, диагностика и лечение / Стуклов Н.И., Альпидовский В.К., Огурцов П.П. – М.: ООО «Медицинское информационное агентство», 2013. – 264 с.
Копирование и тиражирование материалов без указания авторов запрещено и преследуется по закону.
Миелодиспластический синдром (МДС) объединяет группу приобретенных заболеваний кроветворной системы, при которой патологический процесс начинается на уровне полипотентной стволовой клетки и обнаруживает себя нарушением пролиферации и дифференцировки клеток одного, двух или трех ростков кроветворения с их последующей гибелью в костном мозге (неэффективный эритропоэз).
В отличие от АА, стволовые клетки присутствуют в костном мозге больных МДС, хотя они функционально неполноценны. Костный мозг при МДС чаще бывает гиперклеточным, нормоклеточным и реже – гипоклеточным, тогда как в периферической крови обнаруживается рефрактерная анемия, нередко лейко- и/или тромбоцитопения.
В основе функциональной патологии полипотентных стволовых клеток лежат хромосомные изменения, которые обнаруживаются у большинства больных МДС. Они имеют клональный характер, аналогичный цитогенетическим изменениям при лейкозах. Хромосомные изменения при МДС разнообразны и включают транслокацию, инверсию и делецию хромосом. К наиболее характерным относятся: трисомия 8, моносомия 5, моносомия 7, делеция Y-хромосомы, делеция длинного плеча 7 (7q-), 11 (11q-), 13 (13q-), 20 (20q-), а также транслокации t(1;3), t(5;7), t(2;11), t(6;9), t(11;27), инверсия 3 хромосомы. У 20% больных наблюдаются множественные нарушения. Часто встречается делеция длинного плеча хромосомы 5 (у 30% больных). Причем установлено, что с этим плечом 5 хромосомы утрачиваются гены, отвечающие за синтез многих ростковых факторов, в том числе гранулоцитарно-макрофагального, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6 и многих других биологически активных веществ, регулирующих кроветворение.
Форма с подобной хромосомной патологией была даже выделена среди больных МДС в 5q-синдром, который чаще встречается у женщин, характеризуется рефрактерной мегалобластной анемией и редко трансформируется в острый лейкоз (менее 5% больных).
Причины, вызывающие хромосомную патологию, неясны. В ряде случаев предполагается действие таких мутагенных факторов, как ионизирующая радиация, действие химических и лекарственных факторов.
Возникшая в костном мозге в одной полипотентной стволовой клетке цитогенетическая патология, обуславливающая в дальнейшем развитие МДС, способна воспроизводиться в потомках смутировавшей стволовой клетки, формируя таким образом патологический клон, клетки которого не способны к нормальной пролиферации и дифференцировке, что внешне проявляется их морфологической дисплазией и последующей костномозговой гибелью (неэффективный эритропоэз). Установлено, что 75% костного мозга при МДС имеют CD95, маркер запрограммированной клеточной гибели – апоптоза. Это обуславливает различные типы цитопений в периферической крови больных МДС.
Заболеваемость МДС составляет 3 – 15 случаев на 100000 населения и частота его повышается до 30 случаев у людей старше 70 лет и 70 случаев – в возрасте старше 80 лет. Средний возраст больных – 60 – 65 лет, у детей МДС встречается крайне редко.
Клиника
Клиническая картина МДС не имеет специфических особенностей. Основные симптомы зависят от глубины и сочетания поражения ростков кроветворения. Основным признаком болезни является рефрактерный анемический синдром, проявляющийся нарастающей слабостью, повышенной утомляемостью и другими свойственными анемии симптомами. У больных МДС с лейкопенией нередко возникают инфекционные осложнения (бронхиты, пневмонии идр.). Геморрагический синдром вследствие тромбоцитопении наблюдается у 10 – 30% больных, и проявляется кровоизлияниями на коже и видимых слизистых, кровоточивостью десен и носовыми кровотечениями.
Какой – либо характерной органной патологии при МДС нет: периферические лимфоузлы, печень и селезенка не увеличены.
Лабораторные данные.
Анемия различной степени выраженности наблюдается практически у всех больных МДС и чаще носит макроцитарный характер. Очень редко наблюдается гипохромия эритроцитов. Нередко присутствуют эллиптоциты, стоматоциты и акантоциты, а также базофильная пунктация и тельца Жолли в эритроцитах. В крови могут присутствовать ядросодержащие клетки красного ряда. Количество ретикулоцитов чаще сниженное.
Часто у больных в анализах крови имеется стойкая нейтропения, причем для гранулоцитов характерно наличие псевдопельгеровской аномалии (лейкоциты с двудольчатыми ядрами и дегрануляцией цитоплазмы).
Тромбоцитопения встречается у половины больных МДС. Среди тромбоцитов встречаются гигантские и дегранулированные формы.
У части больных МДС в анализах крови могут встречаться бластные клетки.
Костный мозг при МДС обычно гиперклеточный, но может быть нормоклеточным, а в редких случаях – даже гипоклеточным. Однако, всегда присутствуют черты дисэритропоэза: мегалобластоидность, многоядерность эритробластов, наличие митозов, патологических делений и ядерных аномалий, мостиков между ними, базофильная пунктация и вакуолизация цитоплазмы. У части больных в костном мозге повышено содержание сидеробластов с кольцевым расположением гранул железа вокруг ядра клетки.
Нарушение дифференцировки предшественников эритроцитов при МДС проявляется повышенным содержанием в них HbF (уровень которого в зрелых эритроцитах нормальный) и наличием в эритробластах пероксидазы и щелочной фосфатазы, что является характерным для нейтрофилов.
Дисгранулоцитопоэз в костном мозге проявляется задержкой созревания гранулоцитов на уровне миелоцитов, нарушением процесса грануляции цитоплазмы и снижением активности щелочной фосфатазы, что свидетельствует об их функциональной неполноценности, часто встречается гипо – или гиперсегментация ядер нейтрофилов.
Дисмегакариоцитопоэз характеризуется преобладанием микроформ и нарушенной отшнуровкой тромбоцитов.
При некоторых формах МДС выявляется повышенное содержание в костном мозге бластных клеток (от 5 до 20%).
При гистологическом исследовании костного мозга, полученного методом трепанобиопсии, у ряда больных имеет место повышенное образование ретикулиновых волокон, причем резко выраженный миелофиброз наблюдается у 10 – 15% больных МДС. Этому варианту МДС, характеризующемуся более выраженной гиперплазией и дисплазией клеток мегакариоцитарного ростка, с почти 100% наличием хромосомных аномалий, свойственны более выраженная анемия, тромбоцитопения и относительно короткая продолжительность жизни больных (медиана выживаемости 9 – 10 мес.).
Диагностика МДС основывается на наличии рефрактерной анемии, устойчивой к терапии витамином B12, фолиевой кислотой, железом и другими гематиками, которая нередко сочетается с нейтро- и тромбоцитопенией и наличием в пунктате костного мозга морфологических признаков дисгематопоэза (нарушения созревания кроветворных клеток).
Классификация МДС:
В настоящее время в клинической практике используются две классификации: Франко-американо-британской группы (FAB) 1982 года и Всемирной организации здравоохранения (ВОЗ) 2008 годам.
Дифференциальный диагноз
РА чаще всего приходится дифференцировать от витамин-B12- и фолиево-дефицитной анемий, при которых также имеется мегалобластное кроветворение и морфологические признаки дисплазии клеток красного ростка, свидетельствующие о неэффективном эритропоэзе. Быстрые клинический и гематологический ответы на терапию витамином B12 или фолиевой кислотой указывают на причинную взаимосвязь между анемией и дефицитом этих витаминов.
РАКС необходимо дифференцировать с приобретенной сидеробластной анемией, обусловленной хронической свинцовой интоксикацией. РЦМД, при которой имеется панцитопения в периферической крови, напоминает апластическую анемию. Наличие нормальной клеточности костного мозга с морфологическими признаками дисмиелопоэза позволяет правильно верифицировать диагноз.
Классификация МДС (ВОЗ, 2008)
Нозологическая форма МДС | Изменения в крови | Изменения в костном мозге |
Рефрактерная анемия (РА) | — анемия — бласты < 1% — моноциты < 1 х 109/л | — дисплазия кроветворения < 10% в одном ростке кроветворения — бласты < 5% — кольцевые сидеробласты < 15% |
Рефрактерная нейтропения (РН) | — нейтропения — бласты < 1% — моноциты < 1 х 109/л | |
Рефрактерная тромбоцитопения (РТ) | — тромбоцитопения — бласты < 1% — моноциты < 1 х 109/л | |
Рефрактерная анемия с кольцевыми сидеробластами (РАКС) | — анемия — бласты < 1% — моноциты < 1 х 109/л | — дисплазия кроветворения. — бласты < 5% — кольцевые сидеробласты > 15% |
Рефрактерная цитопения с многоростковой дисплазией (РЦМД) | — цитопения по 2 – 3 росткам — бласты < 1% — моноциты < 1 х 109/л | — дисплазия кроветворения < 10% в двух и более ростках кроветворения — бласты < 5% — кольцевые сидеробласты (любое количество) |
Рефрактерная анемия с избытком бластов I (РАИБ-1) | — цитопения любая — бласты < 5% — моноциты < 1 х 109/л | — множественная дисплазия во всех ростках кроветворения — бласты 5 – 9% |
Рефрактерная анемия с избытком бластов II (РАИБ-2) | — цитопения любая — бласты 5 – 19% — моноциты < 1 х 109/л | — множественная дисплазия во всех ростках кроветворения — бласты 10 – 19% — палочки Ауэра ± |
МДС неклассифици-рованный (МДС-Н) | — цитопения любая — бласты <1% | — дисплазия кроветворения < 10% в одном или несколь- ких ростках кроветворения — бласты < 5% |
Синдром 5q- | — анемия — бласты < 1% — тромбоциты норма или увеличены | — нормальное или увеличенное количество мегакариоцитов с гипосегментированными ядрами — изолированная делеция 5q — бласты < 5% |
Гипопластический вариант МДС отличить от АА значительно труднее. В пользу гипоплазии при МДС говорит наличие хромосомной патологии, отсутствующей при АА, высокое содержание на гемопоэтических клетках проапоптических белков (CD95) и низкий уровень щелочной фосфатазы в гранулоцитах при МДС в отличие от нормального содержания этого фермента при АА.МДС с избытком бластов отличается от острого лейкоза по количественному содержанию бластных клеток в костном мозге: все случаи с бластозом более 20% рассматриваются как острый лейкоз.
Лечение
Симптоматическая терапия
Ведущее место в лечении МДС занимает поддерживающая терапия, в первую очередь – переливание эритроцитарной массы, сопровождающееся введением десферала или деферазирокса для удаления избытка железа. Переливание эритроцитарной массы показано при снижении уровня Hb до 80 г/л и ниже, а частота ее зависит от динамики показателей красной крови. Для борьбы с геморрагическим диатезом используется введение тромбоконцентрата, показания те же, что и при лечении АА. При инфекционных осложнениях, обусловленных гранулоцитопенией, показано введение антибиотиков.
Патогенетическая терапия зависит от количества бластов к костном мозге. При выраженом бластозе (> 10%) необходимо регулярно проводить стернальные пункции, чтобы исключить трансформацию МДС в острый лейкоз (acuteleukemia, AL). При увеличении бластов больше 20% терапия проводится по программам лечения AL.
Алгоритм лечения МДС (Савченко В.Г., Кохно А.В., Паровичникова Е.Н.)
Клеточность костного мозга | |||
Гипоклеточный костный мозг | Нормо/гиперклеточный костный мозг | ||
<5% бластов | 5 – 20% бластов | <5% бластов | 5 – 20% бластов |
СуА | СуА | рчЭПО | Децитабин, азацитидин |
АТГ | АТГ | Спленэктомия | FLAG, 7 + 3 |
Спленэктомия | Децитабин, азацитидин | Интерферон-α | МДЦ – 14 дней |
рчЭПО | МДЦ – 14 дней, 6 – МП, мельфалан | Децитабин, азацитидин | 6 – МП |
В случаях количества бластов в костном мозге стойко ниже 20% для принятия решения о тактике лечения необходимо проведение трепанобиопсии, которая позволяет установить клеточность костного мозга. После чего терапия МДС может быть направлена на стимулирование кроветворения при гипоплазии костного мозга (рекомбинантный человеческий эритропоэтин – рч-ЭПО), иммуносупрессию с целью активации стволовых клеток (АТГ, CyA), снижение гемолиза и секвестрации клеток крови (спленэктомия). При гиперклеточных вариантах или формах МДС с бластозом более 5% лечение должно включать подавление опухолевого роста (химиотерапию). В России наиболее подходящий алгоритм выбора терапии МДС, схема которого указана в таблице, сформулирован специалистами Гематологического научного центра: Савченко В.Г., Кохно А.В., Паровичниковой Е.Н. в 2012 году.
В последние годы для стимуляции эритропоэза у больных МДС, иногда успешно, используется рчЭПО: рекормон, эритростим, эпрекс, аранесп и др., который особенно эффективен при низкой концентрации в крови нативного ЭПО (< 500 ед/мл). РчЭПО рекомендуется применять в дозе 100000 МЕ 3 раза в неделю подкожно или по 30000 – 40000 МЕ раз в неделю (при использовании пролонгированных форм эритропоэтина). Терапия считается эффективной при приросте гемоглобина более чем на 10 г/л за 4 – 8 недель или снижение зависимости от гемотрансфузий. Целевая концентрация гемоглобина 120 г/л. Через 2 месяца лечения рчЭПО сообщается о положительном эффекте у 41,6% больных с РА и у 76% больных с РАКС, причем к 6 месяцу этот эффект сохраняется соответственно у 33% и 58%. Таким образом, наиболее эффективным применение ЭПО оказалось у больных при варианте МДС-РАКС.
У белее чем трети больных МДС тяжесть тромбоцитопении может быть временно снижена введением интерферона-α, это позволяет избежать аллоиммунизации, обусловленной введением тромбоконцентрата.Терапия глюкокортикоидами при МДС не эффективна, хотя иногда может уменьшить интенсивность геморрагического синдрома.
У больных МДС с гипопластической фазой заболевания, как и при АА, эффективным оказалось проведение иммуносупрессивной терапии (СуА), которая не только блокирует действие Т-клеток-супрессоров, но и ингибирует клеточный апоптоз. Циклоспорин А назначается в дозе 5 мг/кг и вызывает гематологическое улучшение у 60больных этой группы (полные ремиссии развиваются реже, частичное улучшение – чаще).
Для лечения форм МДС РА, РАКС, РЦМД в качестве первичного метода лечения у пожилых (старше 60 лет) больных с гипоплазией кроветворения или при резистентности к циклоспорину в настоящее время широко применяется спленэктомия с биопсией печении. Наряду с лечебным эффектом данный подход позволяет исключить другие причины развития дисплазии кроветворения. Как правил, спленэктомия позволяет добиться длительных перерывов в гемотрансфузиях, улучшить качество жизни больных.
Использование цитостатических препаратов при РАИБ-варианте МДС в настоящее время считается самым эффективным лечением. До недавнего времени в качестве патогенетической терапии применяли в основном малые дозы цитозара и мелфалан. Схема лечения малыми дозами цитозара выглядит следующим образом. Вводят подкожно по 10 мг/м2 2 раза в день в течение 14, 21 или 28 дней в зависимости от количества бластов и клеточности костного мозга. Мельфалан применяют в дозах 5 – 10 мг/м2 в течение 5 дней peros. Такие курсы проводят раз в месяц, как правило, отполугода до 3 лет, с оценкой терапевтического эффекта каждые 2 – 4 месяца. Эффективной терапия считается при нормализации или относительной нормализации показателей периферической крови и костного мозга, при отсутствии или резком снижении зависимости от гемотрансфузий. Использование указанных схем лечения приводит к развитию частичной ремиссии у 56% больных. Однако, на выживаемость больных такая терапия существенно не влияет.
При тяжелом состоянии больных и невозможности проведения адекватной терапии при МДС-РАИБ-1 и -2 возможно назначать 6-меркаптопурин по 60 мг/м2 в сутки peros в течение 3 лет.
В настоящее время делаются попытки использовать в лечении МДС талидомида и его аналога леналидомида, лишенного нейтротоксической активности, но являющегося мощным ингибитором протеаз. Применение леналидомида вызвало снижение трансфузионной зависимости у 67% больных, причем у 58% достигалась полная независимость от трансфузионной терапии. Стоит отметить, что этот препарат особенно эффективен при 5q-варианте МДС, где его эффективность равна 91%, тогда как при других нарушениях кариотипа – только 19%.
У молодых больных до 60 лет в стандарты лечения МДС-РАИБ-2 входит полихимиотерапия. Используют курсы, применяемые в лечении острых миелобластных лейкозах: «7 + 3» и «FLAG». «7 + 3»: цитарабин 100 мг/м2 в/в капельно каждые 12 часов 1 – 7 дни курса и идарубицин 12 мг/м2 в/в капельно 1 – 3 дни курса. «FLAG»: флударабин 25 мг/м2 в/в капельно 1 – 5 дни курса, цитарабин 2 г/м2 в/в капельно 1 – 5 дни курса + Г-КСФ (гранулоцитарный колониестимулирующий фактор) 5 мкг/кг п/к ежедневно до выхода из цитопении.
Из других активно разрабатываемых препаратов в гематологической практике заслуживают внимание триоксид мышьяка, бевацизумаб (авастин) и др.
В последнее время в клиническую практику внедрены современные цитостатические препараты ингибиторы ДНК-метилтрансфераз. Механизм их действия связан с ингибированием процесса метилирования ДНК в клетках опухолевого клона, что приводит к повышению активности генов, регулирующих клеточный цикл и нормализации процессов дифференцировки клеток костного мозга. Два основных вещества зарегестрированы в России под названием децитабин (Дакоген), азацитидин (Ведаза). По опубликованным данным крупнейших международных исследований эффективность использования этих препаратов в лечении МДС составила 50 – 70%. Децитабин вводят в дозе 20 мг/м2 в/в капельно 1 – 5 дни раз в месяц. Таких курсов проводят4, далее оценивают эффект. При положительной оценке продолжают терапию в течение долгого времени до развития осложнений, при отсутствии эффекта используют другие препараты. Азацитидин вводят подкожно 75 мг/м2 1 – 7 дни раз в месяц. Оценивают эффект через полгода, далее или продолжают терапию длительно или меняют препараты.
Необходимо знать, что самым серьезным осложнением химиотерапии, требующим иногда отмены лечения, является цитопения. Цитопения, как правило, проявляется снижением всех показателей крови (Hb, лейкоциты и тромбоциты). Тяжелыми состояниями, угрожающими жизни считается анемия менее 70 г/л, тромбоцитопения менее 20 х 109/л, лейкопения менее 1 х 109/л или нейтропения менее 0,5 х 109/л. Такие состояния требуют обязательного стационарного лечения, проведения трансфузионной и антибактериальной терапии.
Единственным радикальным методом лечения МДС могла бы стать аллогенная трансплантация костного мозга, однако, применение этого метода ограничивается пожилым возрастом больных, подавляющее большинство которых старше 60 лет.
Прогноз при МДС остается неблагоприятным и зависит от варианта МДС. При РА трансформация в острый лейкоз наблюдается у 15% больных, а медиана выживаемости составляет 50 месяцев. При РАКС эти показатели составляют соответственно 8% и 51 месяц; при РАИБ – 44% и 11 месяцев.
Источник