Метаболический синдром у женщин патофизиология и клиника

Большинство авторов сходятся во мнении о существовании нескольких механизмов, обусловливающих наличие связи артериальной гипертензии и инсулинорезистентности.
Еще в 80-х годах прошлого века ученые пришли к выводу, что сочетание артериальной гипертензии с метаболическими факторами риска – это не механическое скопление, а закономерное проявление единой цепи целого ряда сложных биохимических нарушений на тканевом уровне. В 1985 г. Было высказано предположение, что гиперинсулинемия может служить связывающим звеном между артериальной гипертензией, ожирением и нарушением толерантности к глюкозе (НТГ). В ряде исследований по прямому определению инсулинорезистентности было показано, что больные с артериальной гипертензией в среднем утилизируют на 40% меньше глюкозы, чем лица с нормальным артериальным давлением.
В эпидемиологических исследованиях продемонстрировано также, что 64% больных с артериальной гипертензией имели инсулинорезистентности и только у половины пациентов она клинически манифестировала с нарушением углеводного обмена. С другой стороны, у 36% больных, имевших гиперлипопротеидемию (ГЛП) или избыточную массу тела (ИМТ), не было выявлено инсулинорезистентности. Таким образом, даже на фоне имеющегося в настоящее время огромного интереса к метаболическому синдрому было бы ошибочным связывать каждый случай эссенциальной артериальной гипертензии с проявлениями тканевой инсулинорезистентности.
Хроническая гиперинсулинемия как проявление тканевой инсулинорезистентности способствует задержке в организме натрия путем ускорения его реабсорбции, что приводит к увеличению объема жидкости и общего периферического сосудистого сопротивления. Повышение активности Na-K-, H- и Ca-Mg-АТФазы под непосредственным воздействием инсулина вызывает увеличение содержания внутриклеточного натрия и кальция, что способствует вазоконстрикции гладкой мускулатуры сосудов. При этом усиливается и чувствительность сосудов к прессорным агентам, таким как адреналин и ангиотензин.
Гиперинсулинемия также способствует активации симпатической нервной системы (СНС), в результате чего возрастает сердечный выброс и стимулируется вазоконстрикция периферических кровеносных сосудов. Симпатическая стимуляция почек запускает мощный механизм развития артериальной гипертензии – ренин-ангиотензин-альдостероновую систему. Исследования показывают, что при сочетании артериальной гипертензии с инсулинорезистентностью активность АПФ является достоверно более высокой по сравнению с больными артериальной гипертензией без проявлений инсулинорезистентности. Ангиотензин 11 – главный действующий компонент ренин-ангиотензин-альдостероновой системы – прямо и косвенно (опосредованно через активацию симпатической нервной системы) повышает давление в клубочковом аппарате, вызывает пролиферацию гладкомышечных стенок артерий, гипертрофию кардиомиоцитов и нарушает функцию эндотелия, что способствует системной артериальной и венозной вазоконстрикции.
Особую роль в ассоциации артериальной гипертензии и инсулинорезистентности играет ожирение абдоминального типа, характерное для метаболиского синдрома. В адипоцитах брыжейки и сальника идет синтез метаболически активных веществ, ингибирующих выработку эндогенного оксида азота, соответственно стимулируя вазоконстрикцию. В последние годы также активно обсуждается роль лептина в усилении активности симпатической нервной системы. Артериальная гипертензия развивается примерно у 60% больных ожирением.
В последнее десятилетие получило развитие учение о роли функции эндотелия в формировании и прогрессировании артериальной гипертензии. Показано, что в патогенезе артериальной гипертензии, связанном с метаболическими нарушениями, эндотелиальная функция является интегральным аспектом синдрома инсулинорезистентности и способствует ее углублению, увеличению реактивности сосудов и дальнейшему формированию артериальной гипертензии.
Источник
ГЛАВА 2. ПАТОФИЗИОЛОГИЯ МЕТАБОЛИЧЕСКОГО СИНДРОМА
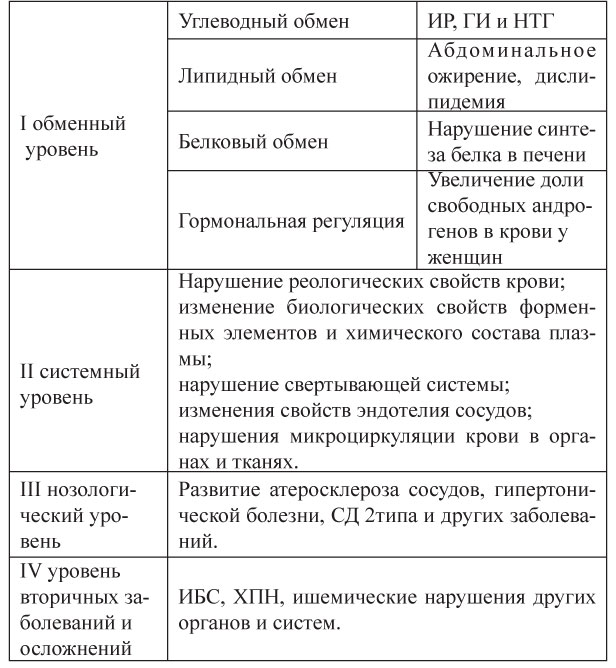
При изучении компонентов МС выделяют несколько уровней (табл.1). Основные пути метаболизма углеводов, жиров и белков тесно взаимосвязаны на уровне узловых метаболитов и ключевых ферментов [4;11].
Таблица 1. Уровни развития нарушений при метаболическом синдроме
Дискоординация метаболизма является первоосновой всех нарушений и основывается на существовании определенных ограничений во взаимных превращениях углеводов, жиров и белков, а именно: ограниченные обратные превращения из углеводов жиры, за счет глицерина; использование углеродного скелета, по крайней мере, 3/4 аминокислот (в том числе незаменимых) для глюконеогенеза и вовлечение углеводных структур в биосинтез лишь заменимых аминокислот; способности аминокислот свой углеродный скелет превращать частично или полностью в ацетил-КоА и, таким образом, служить материалом для синтеза жирных кислот. Эти ограничения усугубляются при инсулиновой недостаточности за счет изменения активности ряда ключевых ферментов обмена веществ, катализирующих фосфорилирова-ние глюкозы и фруктозо-6-фосфата, синтез гликогена из УДФ-1-глюкозы, фосфоролиз гликогена до глюкозо-1-фосфата, дефосфо-рилирование глюкозо-6-фосфата путем гидролиза до свободной глюкозы, превращение аминокислот в α-кетокислоты с помощью реакций переаминирования и окислительного дезаминирования, обратное превращение пировиноградной кислоты в фосфоенол-пируват, липолиз триглицеридов, образование ацетоновых тел из ацетил-КоА.
Помимо регуляторов, вмешивающихся в метаболические процессы на уровне ферментных реакций, существует влияние гормонов, связанное с их выбросом в кровеносное русло. Так, адреналин и норадреналин увеличивают скорость липолиза в жировой ткани за счет стимуляции аденилатциклазы адипоцитов и синтеза цАМФ. Действие глюкагона сходно с действием кате-холаминов. Инсулин оказывает противоположное адреналину и глюкагону действие на липолиз и мобилизацию жирных кислот. СТГ, АКТГ также оказывают стимулирующее влияние на липолиз, увеличивая содержание жирных кислот в плазме крови [11].
Жировая ткань обладает ауто-, пара- и эндокринной функцией и секретирует «адипоцитокины», обладающие различным биологическим действием, которые могут (при их избытке — ожирении), вызывать развитие сопутствующих ожирению осложнений, в том числе ИР: лептин, фактор некроза опухоли-а (TNF—а), ингибитор-1 активатора плазминогена (PAI), протеин, стимулирующий ацилирование (ASK), интерлейкин-6, интер-лейкин-8, ангиотензин-П, резистин, адипонектин, адипсин, протеин agouti, трансформирующий фактор роста-β, адипофилин [4; 11;41]. Многие исследователи рассматривают TNF-a, как медиатор ИР при ожирении. TNF-a снижает активность тирозинкиназы инсулинового рецептора, тормозит экспрессию внутриклеточных переносчиков глюкозы ГЛЮТ-4 в мышечной и жировой ткани. Как показано in vivo, TNF-a может действовать в синергизме с интерлейкинами-1 и 6, а также стимулировать секрецию лептина [33].
Лептин — «голос» жировой ткани, регулирует пищевое поведение, воздействуя на центр насыщения в гипоталамусе. К физиологическим эффектам лептина относятся: повышение тонуса симпатической нервной системы, усиление термогенеза в адипоцитах, снижение синтеза инсулина, снижение транспорта глюкозы, воздействуя на инсулиновый рецептор клетки. Выявлено стимулирующее действие лептина на секрецию гонадотро-пинов. В препубертатном периоде уровень лептина параллельно повышается с увеличением массы до максимальных значений с началом полового созревания. В пубертатном периоде повышается чувствительность к лептину. Ожирение может быть связано с дефицитом лептина и лептинорезистентностью [41]. Рецепторы лептина присутствуют и в яичниках, причем непосредственное влияние на стероидогенез в яичниках может быть как стимулирующим, так и ингибирующим (в эксперементах на животных есть данные о снижении инсулинзависимого синтеза прогестерона и Е2 в клетках гранулезы). Выявлено, что в течение менструального цикла уровень лептина постепенно нарастает на протяжении фолликулиновой фазы, достигая пика в лютеиновую фазу [10;11; 19; 27; 47].
Количество инсулина и лептина в циркуляторном русле прямо пропорционально массе жировых отложений, и их называют «сигналами ожирения» Повышенный уровень лептина при лептинорезистентности и МС обусловливает развитие гормональной дисфункции и висцерального ожирения. Глюкокор-тикоидная нестабильность (внутриклеточный гиперкортицизм) при метаболическом синдроме и инсулинорезистентности так же приводит к развитию висцерального ожирения [51].
В транспорте половых стероидов активную роль играет циркулирующий ГСПС (глобулин, связывающий половые стероиды, или тестостерон-эстрадиол связывающий глобулин). Установлено, что количество ГСПС определяется как наследственными факторами, так и наличием некоторой экстрагенитальной и генитальной патологии. Наличие положительной корреляции между уровнем ГСПС и ХС ЛПВП и обратной между ГСПС и ХС ЛПНП и ЛПОНП обуславливает наследование данного глобулина как генетического фактора риска развития атеросклероза сосудов головного мозга, ИБС и АГ [19]. Установлена прямая корреляция между содержанием эстрона, 17β-эстрадиола и индексом массы тела, обратная корреляция существует между последним показателем и уровнем ГСПС в сыворотке крови, что особенно характерно для пациенток в периоде постменопаузы [38]. Снижение уровня ГСПС в постменопаузе приводит к росту концентрации свободного тестостерона, относительной гиперандрогении и вносит определенный вклад в формирование абдоминального ожирения. Отмечено, что у женщин с гиноидным типом распределения жировой клетчатки уровень ГСПС выше, чем у таковых с андро-идным типом [48].
При исследовании сыворотки крови пациенток, страдающих сахарным диабетом, было установлено, что нарастание уровня глюкозы и повышение уровня инсулина ведут к увеличению свободного тестостерона и понижению уровня ГСПС [11]. Есть данные о том, что у женщин с анамнестически ранним менархе (до 13 лет), в репродуктивном возрасте уровень ГСПС в сыворотке крови более низкий по сравнению с женщинами с поздним менархе [2].
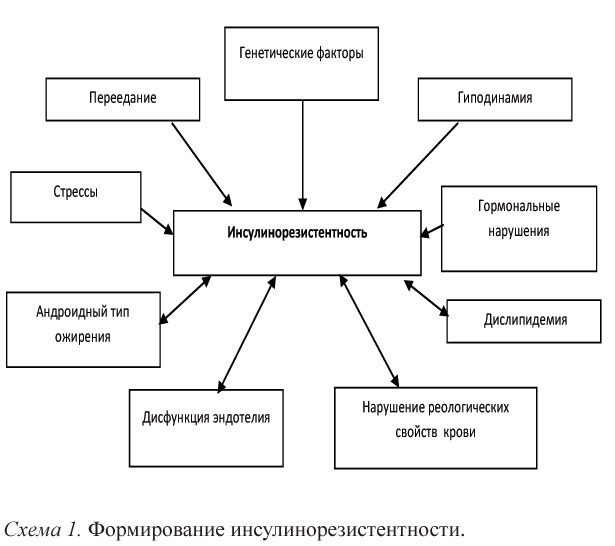
Ключевым моментом в первичных метаболических нарушениях является формирование инсулинорезистентности. Под ИР в настоящее время понимают первичное, селективное и специфическое нарушение биологического действия инсулина, сопровождающееся снижением потребления глюкозы тканями (преимущественно скелетными мышцами) и приводящее к хронической ГИ. Селективный характер ИР означает, что отдельные эффекты инсулина сохраняются, например, реабсорбция натрия в почечных канальцах или влияние на симпатический отдел нервной системы (схема 1).
Практически все составляющие МС являются факторами риска развития сердечно-сосудистых заболеваний, а в сочетании многократно ускоряют их развитие. Причем сочетания отдельных компонентов могут рассматриваться в рамках МС только при наличии ИР. Разумеется, не все компоненты метаболического синдрома встречаются одновременно. Каким фенотипом проявится метаболический синдром, зависит от взаимодействия факторов генетических и внешней среды.
Патогенез дислипидемии при метаболическом синдроме
Наиболее частым вариантом дислипидемии при метаболическом синдроме является липидная триада: сочетание гипер-триглицеридемии, низкого уровня ХС ЛПВП и повышения фракции мелких плотных частиц ХС ЛПОНП, переносчиков тригли-церидов, что является результатом их повышенной печеночной продукции и сниженной элиминации. Механизм влияния ИР на развитие липидных нарушений представлены на схеме 2.
Гиперинсулинемия способствует увеличению пролиферации гладкомышечных клеток и фибропластов, увеличению активности рецепторов ХС ЛПНП и синтезу эндогенного ХС в клетках сосудистой стенки, коллагена, стимуляции выработки ИПФР.
Патогенез развития артериальной гипертензии при метаболическом синдроме
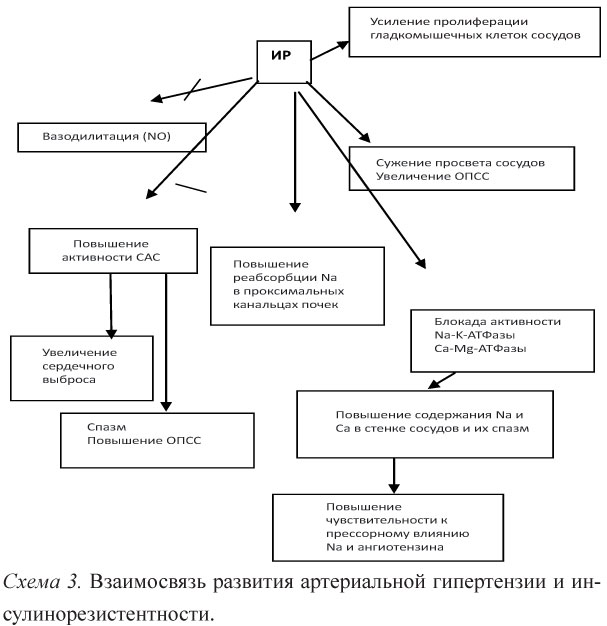
Механизм развития АГ при МС многогранен и неоднозначен. Общее влияние инсулина на АД представляет собой равновесие между прямым вазодилататорным и непрямым вазокон-стрикторным эффектами. При хронической ГИ возникает парадоксальная реакция со стороны сосудов в связи с преобладанием митогенного и симпатикостимулирующего компонентов (схема 3).
С другой сторон, возможен механизм, посредством которого АГ может сама способствовать ИР. Увеличение активности симпатической нервной системы, возникающей при АГ, вызывает понижение объемного кровотока в капиллярах скелетной мускулатуры в результате их вазоконстрикции, что увеличение пути диффузии глюкозы к клеткам и приводит к инсулинорезистент-ности.
Эндотелий сосудов обладает метаболической и секреторной активностью и играет ключевую роль в регуляции тонуса и проницаемости сосудов. Уникальное положение клеток эндотелия на границе между циркулирующей кровью и тканями делает их наиболее уязвимыми для различных патогенных факторов, находящихся в системном и тканевом кровотоке.
В настоящее время есть две основные точки зрения относительно формирования эндотелиопатии. Первая, что при синдроме ИР развивается дисфункция эндотелия сосудов и, в частности, нарушается синтез оксида азота в сосудистой стенке (оксид азота является мощным вазодилататором). Он оказывает сдерживающее влияние на пролиферацию гладкомышечных клеток, тормозит адгезию моноцитов к эндотелию сосудистой стенки, снижает перекисное окисление липидов, т.е. предохраняет стенки сосудов от повреждения. Существует также мнение, что дисфункция эндотелия является не следствием, а причиной в развитии ИР, одним из первичных дефектов, лежащих в основе ее развития. В случае первичного дефекта эндотелиальных клеток трансэндотелиальный транспорт инсулина нарушается, что может способствовать развитию ИР. Однако, до настоящего времени не получено достаточно данных в пользу первичной или вторичной роли эндотелиопатии в генезе инсулино-резистентности [4; 6]. Итак, основные механизмы повышения АД:
- объемозависимый
- повышение активности ренин-ангиотензин-адьдостеро-новой и симпатоадреналовой систем
- дисфункция эндотелия
- гиперлептинемия
- блокада вазодилатирующего эффекта инсулина.
Имеются данные, указывающие на снижение фибриноли-тической активности у пациентов с МС. PAI-1 является основнымингибитором активатора плазминогена, обеспечивая до 60% общейингибиторной активности в отношении активаторов плазминогена вплазме. Повышение уровня PAI-1 связано с риском тромбозов. PAI-1 синтезируется эндотелиальными клетками, моноцитами, макрофагами, гладкомышечными клетками, адипоцитами висцеральнойжировой ткани. Эндотелиальные клетки и тромбоциты регулируютвыделение PAI-1 в процессе фибринолиза. Было установлено, чтоГИ, гипергликемия и гипертриглицеридемия приводят к значительному повышению экспрессии гена, ответственного за продукциюPAI-1, и, соответственно, к повышению концентрации в крови. Внастоящее время считается, что повышение уровня PAI-1 являетсямаркером высокого риска инфаркта миокарда, ассоциируется ссахарным диабетом [4].
Источник
ПАТОФИЗИОЛОГИЯ ИЗМЕНЕНИЙ ПРИ МЕТАБОЛИЧЕСКОМ СИНДРОМЕ
СОДЕРЖАНИЕ
1. Основные пути метаболизма углеводов, жиров и белков
2. Патогенез дислипидемии при метаболическом синдроме
3. Патогенез развития артериальной гипертензии при метаболическом синдроме
Основные пути метаболизма углеводов, жиров и белков
При изучении компонентов МС выделяют несколько уровней (табл.1). Основные пути метаболизма углеводов, жиров и белков тесно взаимосвязаны на уровне узловых метаболитов и ключевых ферментов
Дискоординация метаболизма является первоосновой всех нарушений и основывается на существовании определенных ограничений во взаимных превращениях углеводов, жиров и белков, а именно: ограниченные обратные превращения из углеводов жиры, за счет глицерина; использование углеродного скелета, по крайней мере, 3/4 аминокислот (в том числе незаменимых) для глюконеогенеза и вовлечение углеводных структур в биосинтез лишь заменимых аминокислот; способности аминокислот свой углеродный скелет превращать частично или полностью в ацетил-КоА и, таким образом, служить материалом для синтеза жирных кислот. Эти ограничения усугубляются при инсулиновой недостаточности за счет изменения активности ряда ключевых ферментов обмена веществ, катализирующих фосфорилирова-ние глюкозы и фруктозо-6-фосфата, синтез гликогена из УДФ-1-глюкозы, фосфоролиз гликогена до глюкозо-1-фосфата, дефосфо-рилирование глюкозо-6-фосфата путем гидролиза до свободной глюкозы, превращение аминокислот в α-кетокислоты с помощью реакций переаминирования и окислительного дезаминирования, обратное превращение пировиноградной кислоты в фосфоенол-пируват, липолиз триглицеридов, образование ацетоновых тел из ацетил-КоА.
Помимо регуляторов, вмешивающихся в метаболические процессы на уровне ферментных реакций, существует влияние гормонов, связанное с их выбросом в кровеносное русло. Так, адреналин и норадреналин увеличивают скорость липолиза в жировой ткани за счет стимуляции аденилатциклазы адипоцитов и синтеза цАМФ. Действие глюкагона сходно с действием кате-холаминов. Инсулин оказывает противоположное адреналину и глюкагону действие на липолиз и мобилизацию жирных кислот. СТГ, АКТГ также оказывают стимулирующее влияние на липолиз, увеличивая содержание жирных кислот в плазме крови [11].
Жировая ткань обладает ауто-, пара- и эндокринной функцией и секретирует «адипоцитокины», обладающие различным биологическим действием, которые могут (при их избытке — ожирении), вызывать развитие сопутствующих ожирению осложнений, в том числе ИР: лептин, фактор некроза опухоли-а (TNF—а), ингибитор-1 активатора плазминогена (PAI), протеин, стимулирующий ацилирование (ASK), интерлейкин-6, интер-лейкин-8, ангиотензин-П, резистин, адипонектин, адипсин, протеин agouti, трансформирующий фактор роста-β, адипофилин [4; 11;41]. Многие исследователи рассматривают TNF-a, как медиатор ИР при ожирении. TNF-a снижает активность тирозинкиназы инсулинового рецептора, тормозит экспрессию внутриклеточных переносчиков глюкозы ГЛЮТ-4 в мышечной и жировой ткани. Как показано in vivo, TNF-a может действовать в синергизме с интерлейкинами-1 и 6, а также стимулировать секрецию лептина [33].
Лептин — «голос» жировой ткани, регулирует пищевое поведение, воздействуя на центр насыщения в гипоталамусе. К физиологическим эффектам лептина относятся: повышение тонуса симпатической нервной системы, усиление термогенеза в адипоцитах, снижение синтеза инсулина, снижение транспорта глюкозы, воздействуя на инсулиновый рецептор клетки. Выявлено стимулирующее действие лептина на секрецию гонадотро-пинов. В препубертатном периоде уровень лептина параллельно повышается с увеличением массы до максимальных значений с началом полового созревания. В пубертатном периоде повышается чувствительность к лептину. Ожирение может быть связано с дефицитом лептина и лептинорезистентностью [41]. Рецепторы лептина присутствуют и в яичниках, причем непосредственное влияние на стероидогенез в яичниках может быть как стимулирующим, так и ингибирующим (в эксперементах на животных есть данные о снижении инсулинзависимого синтеза прогестерона и Е2 в клетках гранулезы). Выявлено, что в течение менструального цикла уровень лептина постепенно нарастает на протяжении фолликулиновой фазы, достигая пика в лютеиновую фазу [10;11; 19; 27; 47].
Количество инсулина и лептина в циркуляторном русле прямо пропорционально массе жировых отложений, и их называют «сигналами ожирения» Повышенный уровень лептина при лептинорезистентности и МС обусловливает развитие гормональной дисфункции и висцерального ожирения. Глюкокор-тикоидная нестабильность (внутриклеточный гиперкортицизм) при метаболическом синдроме и инсулинорезистентности так же приводит к развитию висцерального ожирения [51].
В транспорте половых стероидов активную роль играет циркулирующий ГСПС (глобулин, связывающий половые стероиды, или тестостерон-эстрадиол связывающий глобулин). Установлено, что количество ГСПС определяется как наследственными факторами, так и наличием некоторой экстрагенитальной и генитальной патологии. Наличие положительной корреляции между уровнем ГСПС и ХС ЛПВП и обратной между ГСПС и ХС ЛПНП и ЛПОНП обуславливает наследование данного глобулина как генетического фактора риска развития атеросклероза сосудов головного мозга, ИБС и АГ [19]. Установлена прямая корреляция между содержанием эстрона, 17β-эстрадиола и индексом массы тела, обратная корреляция существует между последним показателем и уровнем ГСПС в сыворотке крови, что особенно характерно для пациенток в периоде постменопаузы [38]. Снижение уровня ГСПС в постменопаузе приводит к росту концентрации свободного тестостерона, относительной гиперандрогении и вносит определенный вклад в формирование абдоминального ожирения. Отмечено, что у женщин с гиноидным типом распределения жировой клетчатки уровень ГСПС выше, чем у таковых с андро-идным типом [48].
При исследовании сыворотки крови пациенток, страдающих сахарным диабетом, было установлено, что нарастание уровня глюкозы и повышение уровня инсулина ведут к увеличению свободного тестостерона и понижению уровня ГСПС [11]. Есть данные о том, что у женщин с анамнестически ранним менархе (до 13 лет), в репродуктивном возрасте уровень ГСПС в сыворотке крови более низкий по сравнению с женщинами с поздним менархе [2].
Ключевым моментом в первичных метаболических нарушениях является формирование инсулинорезистентности. Под ИР в настоящее время понимают первичное, селективное и специфическое нарушение биологического действия инсулина, сопровождающееся снижением потребления глюкозы тканями (преимущественно скелетными мышцами) и приводящее к хронической ГИ. Селективный характер ИР означает, что отдельные эффекты инсулина сохраняются, например, реабсорбция натрия в почечных канальцах или влияние на симпатический отдел нервной системы (схема 1).
Практически все составляющие МС являются факторами риска развития сердечно-сосудистых заболеваний, а в сочетании многократно ускоряют их развитие. Причем сочетания отдельных компонентов могут рассматриваться в рамках МС только при наличии ИР. Разумеется, не все компоненты метаболического синдрома встречаются одновременно. Каким фенотипом проявится метаболический синдром, зависит от взаимодействия факторов генетических и внешней среды.
Патогенез дислипидемии при метаболическом синдроме
Наиболее частым вариантом дислипидемии при метаболическом синдроме является липидная триада: сочетание гипер-триглицеридемии, низкого уровня ХС ЛПВП и повышения фракции мелких плотных частиц ХС ЛПОНП, переносчиков тригли-церидов, что является результатом их повышенной печеночной продукции и сниженной элиминации. Механизм влияния ИР на развитие липидных нарушений представлены на схеме 2.
Гиперинсулинемия способствует увеличению пролиферации гладкомышечных клеток и фибропластов, увеличению активности рецепторов ХС ЛПНП и синтезу эндогенного ХС в клетках сосудистой стенки, коллагена, стимуляции выработки ИПФР.
Патогенез развития артериальной гипертензии при метаболическом синдроме
Механизм развития АГ при МС многогранен и неоднозначен. Общее влияние инсулина на АД представляет собой равновесие между прямым вазодилататорным и непрямым вазокон-стрикторным эффектами. При хронической ГИ возникает парадоксальная реакция со стороны сосудов в связи с преобладанием митогенного и симпатикостимулирующего компонентов (схема 3).
С другой сторон, возможен механизм, посредством которого АГ может сама способствовать ИР. Увеличение активности симпатической нервной системы, возникающей при АГ, вызывает понижение объемного кровотока в капиллярах скелетной мускулатуры в результате их вазоконстрикции, что увеличение пути диффузии глюкозы к клеткам и приводит к инсулинорезистент-ности.
Эндотелий сосудов обладает метаболической и секреторной активностью и играет ключевую роль в регуляции тонуса и проницаемости сосудов. Уникальное положение клеток эндотелия на границе между циркулирующей кровью и тканями делает их наиболее уязвимыми для различных патогенных факторов, находящихся в системном и тканевом кровотоке.
В настоящее время есть две основные точки зрения относительно формирования эндотелиопатии. Первая, что при синдроме ИР развивается дисфункция эндотелия сосудов и, в частности, нарушается синтез оксида азота в сосудистой стенке (оксид азота является мощным вазодилататором). Он оказывает сдерживающее влияние на пролиферацию гладкомышечных клеток, тормозит адгезию моноцитов к эндотелию сосудистой стенки, снижает перекисное окисление липидов, т.е. предохраняет стенки сосудов от повреждения. Существует также мнение, что дисфункция эндотелия является не следствием, а причиной в развитии ИР, одним из первичных дефектов, лежащих в основе ее развития. В случае первичного дефекта эндотелиальных клеток трансэндотелиальный транспорт инсулина нарушается, что может способствовать развитию ИР. Однако, до настоящего времени не получено достаточно данных в пользу первичной или вторичной роли эндотелиопатии в генезе инсулино-резистентности [4; 6]. Итак, основные механизмы повышения АД:
- объемозависимый
- повышение активности ренин-ангиотензин-адьдостеро-новой и симпатоадреналовой систем
- дисфункция эндотелия
- гиперлептинемия
- блокада вазодилатирующего эффекта инсулина.
Имеются данные, указывающие на снижение фибриноли-тической активности у пациентов с МС. PAI-1 является основнымингибитором активатора плазминогена, обеспечивая до 60% общейингибиторной активности в отношении активаторов плазминогена вплазме. Повышение уровня PAI-1 связано с риском тромбозов. PAI-1 синтезируется эндотелиальными клетками, моноцитами, макрофагами, гладкомышечными клетками, адипоцитами висцеральнойжировой ткани. Эндотелиальные клетки и тромбоциты регулируютвыделение PAI-1 в процессе фибринолиза. Было установлено, чтоГИ, гипергликемия и гипертриглицеридемия приводят к значительному повышению экспрессии гена, ответственного за продукциюPAI-1, и, соответственно, к повышению концентрации в крови. Внастоящее время считается, что повышение уровня PAI-1 являетсямаркером высокого риска инфаркта миокарда, ассоциируется ссахарным диабетом.
ЛИТЕРАТУРА:
- Алгоритмы специализированной медицинской помощи больным сахарным диабетом / под ред. И.И. Дедова, М.В. Шестаковой. — М., 2007. — 105 с.
- Алиева Н.А. Особенности репродуктивного здоровья девушек подростков с ожирением различного генеза: Автореф. дис. … канд. мед. наук. — Волгоград. — 2007.- 18 с.
- Белоцерковцева Л.Д., Корнеева Е.В., Ерченко Е.Н. Метаболические нарушения у пациенток с гиперандрогенией и хронической ановуляцией // Материалы V Cибирского физиологического съезда // Бюллетень сибирской медицины. — 2005.- № 4.- С.82.
- Белоцерковцева Л.Д., Коваленко Л.В., Корнеева Е.В., Шиша-нок О.Ю. Перименопауза и метаболический синдром // Вестник СурГУ. Медицина.- 2008. — №1.- С. 40-52.
- Белоцерковцева Л.Д., Коваленко Л.В., Васечко Т.М., Ерченко Е.Н. Состояние углеводного и жирового обмена и риск перинатальной патологии у беременных с ожирением // Вестник новых медицинских технологий. 2008. — № 2. — С. 55-60.
- Беляков Н.А., Сеидова Г.Б., Чубриева С.Ю. и др. Метаболический синдром у женщин (патофизиология и клиника). — СПб.: Издательский дом СПбМАПО. 2005.-440с.
- Бериханова Р.Р., Хрипунова Г.И. Особенности течения родов у пациенток с метаболическим синдромом // Материалы IV съезда акушеров-гинекологов России. — Москва, 2008. — С. 27-28.
- Гинекология. Курс лекций. / Под ред. А.Н.Стрижакова, А.И.Давыдова. — М.: ГЭОТАР-МЕД, 2009. — 472 с.
- Давыдов А.И., Стрижакова М.А., Орлов О.Н. Роль лептина в регуляции репродуктивной системы женщины // Вопросы гинекологии, акушерства и перинатологии.2004. — Т.3, №6. — С.84-89.
- Дедов И.И., Мельниченко Г.А. Ожирение. Этиология, патогенез, клинические аспекты. — М.: Медицинское информационное агентство.2004. — С.216-232.
- Ерченко Е.Н. Патофизиологические особенности углеводного и липидного обменов и состояние новорожденных у беременных с избыточной массой тела и ожирением: Автореф. дис. … канд. мед. наук. — Москва, 2009. — 28 с.
- Кадамалиева М.Д., Абдурахманова Ф.М. Особенности течения беременности, родов и послеродового периода при ожирении // Материалы VIII Всероссийского научного форума «Мать и дитя» — М., 2006. — С.101.
- Кантемирова З.Р., Петухов В.А. Беременность, желчный пузырь и липидный дистресс-синдром: диагностика и принципы лечения // Гинекология. — 2005. — Т. 7. — № 2. — С. 76-79.
- Кисляк О.А. Артериальная гипертензия в подростковом возрасте. — М.: Миклош. — 2007. — 288 с.
- Леонтьева И.В. Метаболический синдром как педиатрическая проблема // Рос. вестник перинатологии и педиатрии. -2008. — Т.53, №3. — С.4-16
- Манухин И.Б., Тумилович Л.Г., Геворкян М.А. Клинические лекции по гинекологической эндокринологии: рук. для врачей. — М.: ГЭОТАР-Медиа, 2006. — 316 с.
- Мельниченко Г.А., Соколова М.Ю., Белова Ю.Ю. Современные подходы к диагностике гестационного диабета// Материалы VIII Всероссийского научного форума «Мать и дитя» — М., 2006. — С. 161-162.
- Овсянникова Т.В., Шешукова Н.А. Возможности заместительной гормональной терапии в зависимости от периода климактерия // Гинекология, 2007. — Т. 9, № 2.
- Овсянникова Т.В. Инновационная терапия климактерических расстройств в постменопаузе //Журнал Российского общества акушеров-гинекологов. — 2009. -№2. -С. 3-5.
- Подзолкова Н.М., Чукарева Н.А., Старцева Т. Гестоз и ожирение: есть проблема — найдено ли решение // Материалы 36-го ежегодного конгресса по изучению патофизиологии беременности и организации гестоза. — Москва, 24-28 мая 2004. — С. 177-178.
- Себко Т.В., Доброхотова Ю.Э., Иванова Т.А., Носиков В.В., Осипова Т.А., Алехин М.В. Генетические маркеры инсулино-резистентности и прогнозирования гестационного сахарного диабета / Материалы X юбилейного Всероссийского научного форума «Мать и дитя» — М., 2009. — С. 402.
- Серов В.Н., Прилепская В.Н., Овсянникова Т.В. Гинекологическая эндокринология. — М.: МЕДпресс-информ, 2004. — 528 с.
- Стрижаков А.Н., Давыдов А.И., И.В. Игнатко, Белоцерков-цева Л.Д. Физиология и патология плода: [монография]. — М.: Медицина, 2004. — 356 с.
- Хейдар Л.А., Бояр Е.А. К вопросу о гестационном сахарном диабете // Медицинский вестник. — 2007. — № 4. — С. 12.
- Чубкин И.В. Метаболический синдром у девушек подросткового возраста: Автореф. дис. … канд. мед. наук.- СПб. — 2007.- 19 с.
- Чубриева С.Ю., Глухов Н.В., Зайчик А.М. Жировая ткань как эндокринный регулятор // Вестник Санкт-Петербургского университета. Серия 11.- 2008
- Шехтман М.М., Варламова Т.М., Бурдули Г.М. Заболевания эндокринной системы и обмена веществ у беременных. — М.: Триада-Х, 2001. — 128 с.
- Шибанова Е.И., Мурашко Л.Е., Дегтярева Е.И. Современные представления об инсулинорезистентности вне и во время беременности // Акушерство и гинекология. — 2009.- № 6. — С. 6 — 9.
- Шилин Д.Е. Коррекция метаболических и эндокринных нарушений при лечении гиперандрогении у девочек и девушек //Архив Фарматека. — 2003. — №16.
Источник