Лимфоматоидный гранулематоз код мкб

Рубрика МКБ-10: C83.8
МКБ-10 / C00-D48 КЛАСС II Новообразования / C00-C97 Злокачественные новообразования / C81-C96 Злокачественные новообразования лимфоидной, кроветворной и родственных им тканей / C83 Диффузная неходжкинская лимфома
Определение и общие сведения[править]
Лимфоматоидный гранулематоз
Лимфоматоидный гранулематоз является редким вирус Эпштейна-Барр — ассоциированным системным ангиодеструктивным лимфопролиферативным заболеванием. Он характеризуется поражением легких, но также может включать в себя некоторые внелегочные поражения (кожа, ЦНС, почки, очень редко лимфатические узлы и селезенка).
Лимфоматоидный гранулематоз был впервые описан в качестве отдельного заболевания в 1972 г.
Эпидемиология
Лимфоматоидный гранулематоз является редким заболеванием. Соотношение мужчины : женщины — 2 : 1. Лимфоматоидный гранулематоз чаще всего встречается в возрасте 40-60 лет.
Этиология и патогенез[править]
Кроме ассоциации с оппортунистической инфекцией и вирусом Эпштейн-Бар, этиология лимфоматоидного гранулематоза неизвестна.
Патогенез
Патогенез лимфоматоидного гранулематоза неизвестен. Однако недавние исследования предоставили убедительные доказательства, что лимфоматоидного гранулематоз является отличительным типом злокачественной лимфомы, связанной с иммуносупрессией.
Клинические проявления[править]
Симптомы зависят от места вовлечения лимфоматоидного гранулематоза, но в основном включают кашель, одышку или боль в груди (при поражение легких) и конституциональные симптомы, такие как потеря веса и лихорадка.
Другие типы диффузных неходжкинских лимфом: Диагностика[править]
Диагноз основан на гистологической триаде, включающей следующее:
1. Узловой полиморфный лимфоидной инфильтрат, состоящий из малых лимфоцитов, плазматических клеток и переменного количества крупных атипичных мононуклеаров
2. Ангиит из-за трансмуральной инфильтрации артерий и вен лимфоцитами (процесс, отличный от васкулита, в котором острые и хронические воспалительные клетки обнаруживаются с расположением в некротическом стенке сосуда)
3. Гранулематоз (центральный некроз в лимфоидных узелках).
Характерных лабораторных отклонений не существуют при лимфоматоидном гранулематозе.
Дифференциальный диагноз[править]
Дифференциальную диагностику проводят с неходжкинской лимфомой и саркоидозом.
Другие типы диффузных неходжкинских лимфом: Лечение[править]
У больных с доброкачественным течением лечение не требуется.
При симптоматическом или прогрессирующем течении, в целом, терапия включает в себя преднизолон с противоопухолевыми препаратами (например, циклофосфамид).
Локализованный процесс может реагировать на лучевую терапию.
Другие варианты лечения включают ганцикловир, интерферон альфа-2, или, в зависимости от гистологического класса, комбинированную химиотерапию.
Прогноз
Средняя продолжительность жизни от момента постановки диагноза составляет 14 месяцев. Более 60% пациентов умирают в течение 5 лет.
Причиной смерти, как правило, является масштабное разрушение легочной паренхимы, что приводит к дыхательной недостаточности, сепсису, и, иногда, к массивному кровохарканью.
Плохие прогностические показатели включают возраст моложе 30 лет, неврологические нарушения или поражение печени, лейкопению или панцитопению.
Профилактика[править]
Прочее[править]
Первичная эффузионная лимфома
Определение и общие сведения
Первичная эффузионная лимфома — это крупноклеточная В-клеточная лимфома, локализующаяся в полостях тела и характеризующаяся лимфоматозными выпотами в плевральную, перитонеальную и перикардиальную полости и всегда ассоциирована с вирусом герпеса человека 8 типа (ВГЧ-8).
Распространенность первичной эффузионной лимфомы неизвестна, считается, что она составляет не менее 1% среди всех не СПИД-ассоциированных лимфом и приблизительно 3% среди СПИД-ассоциированных лимфом.
Этиология и патогенез
Точная этиология первичной эффузионной лимфомы неизвестна. Первичная эффузионная лимфома всегда ассоциирована с ВГЧ-8, который также известен, как герпесвирус, ассоциированный с саркомой Капоши и наиболее часто обнаруживается у иммунодефицитных пациентов, особенно, на развернутой клинической стадии СПИД. Инфицированность вирусом Эпштейн-Барр также часто обнаруживается у большинства больных первичной эффузионной лимфомой. Полагают, что генные продукты этих вирусных геномов ингибируют клеточный апоптоз и способствуют неконтролируемому делению клеток с последующей неопластической трансформацией.
Клинические проявления
Первичная эффузионная лимфома наиболее часто обнаруживается у молодых мужчин, положительных на ВИЧ или имеющих доклиническую стадию СПИДа. Описаны очень редкие не ВИЧ-ассоциированные случаи, но почти все пациенты были пожилого возраста или имели не ВИЧ-ассоциированный иммунодефицит. Перитонеальные, плевральные и перикардиальные лимфоматозные выпоты наблюдались у большинства пациентов без экстранодального распространения лимфомы.
Симптоматика первичной эффузионной лимфомы зависит от полости, в которой локализуется лимфома и определяется клиникой выпота. Плевральные и перикардиальные лимфомы сопровождаются одышкой, в то время как перитонеальная лимфома проявляется увеличением живота. Первичная эффузионная лимфома обычно крайне агрессивна, диссеминация лимфомы в различные локализации, оппортунистические инфекции и ВИЧ-ассоциированные осложнения зачастую фатальны. В редких, ВИЧ-негативных случаях исход болезни может быть благоприятнее.
Диагностика
Цитологический анализ выпота и проточная цитометрия определяют наличие крупноклеточных клональных неопластических клеток, имеющих иммунобластый, анапластический или плазмабластный внешний вид, с заметными ядрышками, с круглыми или неровными ядрами и иногда с вакуолизированной цитоплазмой. Постановка диагноза первичной эффузионной лимфомы требует присутствия ВГЧ-8. Тест на ассоциированный с латентностью ядерный антиген-1 (LANA-1) определяет любой признак присутствия ВГЧ-8 в тканевом образце. Полный анализ крови и позитрон-эмиссионную томографию/компьютерную томографию также следует выполнять для определения распространения лимфомы.
Дифференциальный диагноз
Дифференциальную диагностику первичной эффузионной лимфомы проводят с диффузной крупноклеточной В-клеточной лимфомой, лимфомой Беркитта, которая может проявляться лимфоидными выпотами, анапластической крупноклеточной лимфомой и пиоторакс-ассоциированной лимфомой.
Лечение
Первичная эффузионная лимфома обычно проявляет рефрактерность к общепринятой химиотерапии и принципы ее лечения пока не определены. Высокоактивную антиретровирусную терапию (ВААРТ) следует проводить одновременно с химиотерапией у всех ВИЧ-позитивных больных первичной эффузионной лимфомой. Химиотерапевтический режим: циклофосфамид-доксорубицин-винкристин и преднизолон (CHOP) используется наиболее часто. Разработка таргетной терапии для первичной эффузионной лимфомы продолжается.
Прогноз
Прогноз при первичной эффузионной лимфоме крайне неблагоприятен со средней продолжительностью жизни 3-4 месяца после постановки диагноза. Редко встречающиеся ВИЧ-негативные больные с первичной эффузионной лимфомой могут достичь продолжительной выживаемости после химиотерапии.
Источники (ссылки)[править]
https://emedicine.medscape.com
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Лимфоматоидный гранулематоз. Диагностика лимфоматоидного гранулематоза.
В классификацию ВОЗ лимфоматоидный гранулематоз включен как пролиферативное заболевание из зрелых В-клеток с неопределенным злокачественным потенциалом. Это заболевание встречается нечасто и почти всегда сопровождается поражением легких; часто поражаются другие экстранодальные области (кожа, мозг, почки, печень). Поражение же лимфатических узлов выявляются редко.
Инфильтрат состоит из малых Т-клеток и различных количеств EBV-позитивных В-клеток, иногда полиморфных Часто встречается ангиоцентрический и ангиодеструктивный вариант роста, что может сопровождаться появлением очагов инфаркта. Повреждение сосудов может быть также индуцировано выделением EBV-опосредованных хемокинов.
Лимфоматоидный гранулематоз по степени дифференцировки (Grade) классифицируется следующим образом:
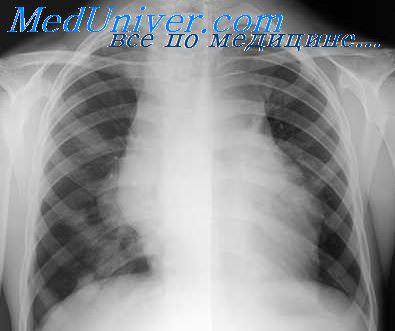
Grade I: при большом увеличении в одном поле зрения менее 5 EBV-позитивных крупных В-клеток; некроз определяется редко. Grade II: при большом увеличении в одном поле зрения 5—20 EBV-позитивных крупных В-клеток; некроз определяется более часто. Grade III: большие количества EBV-позитивных крупных В-клеток, формирующие в некоторых областях сливающиеся пласты; различные количества Т-клеток; частый некроз.
При Grade II и Grade III В-клетки обычно клональные.) Этот подтип следует рассматривать как вариант диффузной В-крупноклеточной лимфомы. Обычно у больных лнмфоматоидным гранулематозом определяется врожденный или приобретенный иммунодефицит. При отсу1ствии же специфических проявлений иммунодефицита у больных (обычно определяются признаки нарушения функции иммуномодуляторов течение заболевания может варьировать.
Лимфоматоидный гранулематоз отличается от полиморфных посттрансплантационных лимфопролиферативных нарушений своими клиническими (преимущественно легочными) проявлениями. Гистологически при полиморфных посттрансплантационных лимфопролиферативных нарушениях определяются В-клетки — от плазмоцитов до иммунобластов, в то время как при лимфоматоидном гранулематозе доминируют малые Т-клстки.
При Т-клеточной/богатой гистиоцитами В-клеточной лимфоме В-клетки могут проявлять EBV-позитивность. Однако обычно эта лимфома сопровождается поражением узлов с отсутствием ангиоинвазивных и ангиодеструктивных признаков лимфоматоидного гранулематоза.
Лимфопролиферативиые заболевания, связанные с первичными иммунодефицитными состояниями, встречаются редко и лежат в основе различных патологических изменений Преимущественно болеют дети, хотя разнообразные иммунодефицитные состояния часто встречаются у взрослых. Первичные иммунные нарушения связаны с полиморфными, поликлональными лимфопролиферативными состояниями, включая лимфоматозный гранулематоз, лейкоз и лимфомы.
Показано, что в большинстве случаев в основе патогенеза лифопролиферативных заболеваний этой группы лежит нарушение Т-клеточного иммунитета по отношению к вирусу Эпштейна-Барр, что приводит к пролиферации В-клеток и, в конечном счете, к формированию опухоли. В большинстве случаев опухоли развиваются вне узлов.
Летальный инфекционный мононуклеоз наблюдается у больных со сцепленным с Х-хромосомой лимфопролиферативным состоянием (синдром Дункана) и тяжелым комбинированным иммунодефицитом (SC1D). EBV-инфекция связана с неудержимым делением EBV-позитивных плазмоцитоидных и иммунобластных клеток в узлах и экстранодально. Смерть может наступить вследствие развития геморрагического синдрома, панцитопении и инфекционных осложнений.
В-крупноклеточная диффузная лимфома и лимфома Беркитта среди всех лимфом встречаются наиболее часто, осложняя первичные иммунодефицитпые заболевания.
Т-клеточный лейкоз/лимфома встречается при атаксической телеажиэктазии (механизм восстановления аномальной ДНК) чаще, чем В-клеточная лимфома.
Лимфома Ходжкина — описаны лишь редкие случаи у больных с первичным иммунодефицитом.
— Также рекомендуем «Лимфоузлы при ВИЧ инфекции. Посттрансплантационные нарушения оттока.»
Оглавление темы «Воспаление и лимфоузлы. Онкология и лимфоузлы. «:
1. Гистология грибовидного микоза. Признаки грибовидного микоза.
2. Т-клеточная лимфома энтеропатического типа. Признаки лимфом энтеропатического типа.
3. Лимфоузлы при ВИЧ инфекции. Посттрансплантационные нарушения оттока.
4. Лимфоматоидный гранулематоз. Диагностика лимфоматоидного гранулематоза.
5. Виды анапластической крупноклеточной лимфомы. Иммуногистология анапластической крупноклеточной лимфомы.
6. Лимфоузлы при ВИЧ инфекции. Посттрансплантационные нарушения оттока.
7. Лимфома Ходжкина. История лимфомы Ходжкина. Признаки болезни ходжкина.
8. Нодулярный тип лимфоидного преобладания лимфомы Ходжкина.
9. Иммунофенотип нодулярной формы болезни Ходжкина. Диагностика нодулярной формы болезни Ходжкина.
10. Классическая лимфома Ходжкина. Признаки лимфомы Ходжкина. Диагнстика лимфомы Ходжкина.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Дифференциальная диагностика
Названия
Название: Лимфоматоидный папулез.

Лимфоматоидный папулез
Описание
Лимфоматоидный папулез. Доброкачественное заболевание лимфатической системы с рецидивирующими полиморфными кожными проявлениями. Клинически сопровождается папулезными высыпаниями различной величины, которые затем трансформируются в буллы, вскрываются, образуя эрозии, корочки, рубцы. Сыпь может располагаться на любом участке кожного покрова, субъективные ощущения отсутствуют. Диагноз выставляется на основании анамнеза, клинических данных, гистологического и иммуногистохимического исследований. В рамках лечебного курса лимфоматоидного папулеза применяются глюкокортикостероиды, антибиотики, цитостатики, ПУВА-терапия, однако все меры дают лишь кратковременный эффект.
Дополнительные факты
Лимфоматоидный папулез – узелковая лимфоидная опухоль кожи доброкачественного характера, имеющая тенденцию к медленному росту. Может быть представлена как единичным образованием, так и множественными элементами. Заболевание не имеет гендерной окраски, сезонности, эндемичности, наследственной предрасположенности. Средний возраст манифестации патологии — 40 лет. Распространенность в популяции составляет 1-2 случая на 1 млн. Человек. У представителей негроидной расы лимфоматоидный папулез практически не встречается. В самостоятельную нозологическую единицу заболевание выделил В. Маколей в 1968 году, отсюда другое название лимфоматоидного папулеза – болезнь Маколея.
В настоящее время лимфоматоидный папулез принято относить к группе первичных CD30+ лимфопролиферативных патологий кожи, наряду с такими процессами, как псевдолимфома, лимфогранулематоз, атипичный регрессирующий гистиоцитоз, анапластическая крупноклеточная лимфома кожи. С перечисленными заболеваниями лимфоматоидный папулез сближает не только общность происхождения, но и нередкое одновременное или последовательное развитие нескольких патологий у одного пациента. Повышенная настороженность дерматологов и онкологов в отношении лимфоматоидного папулеза объясняется возможностью его трансформации в злокачественную Т-клеточную лимфому (в 10-20% случаев).
Лимфоматоидный папулез
Причины
Этиология заболевания неизвестна. Патогенез также является предметом споров и обсуждений. Большинство дерматологов, основываясь на гистологической картине лимфоматоидного папулеза, которая выглядит как истинная лимфома, считают заболевание лимфоцитарной опухолью кожи низкой степени злокачественности. Другие рассматривают лимфоматоидный папулез в качестве ограниченной доброкачественной лимфоплазии, возникающей при участии CD4-лимфоцитов, которые регулируют силу иммунного ответа. По всей вероятности, истина находится где-то посередине.
В развитии лимфоматоидного папулеза играет роль нарушение в дифференцировке Т-лимфоцитов, когда в результате латентно текущих аутоиммунных и системных заболеваний, врождённого или приобретённого иммунодефицита, ионизирующего излучения они начинают мутировать, провоцируя усиленную пролиферацию эпидермальных клеток, как здоровых, так и генетически деструктированных. Параллельно изменяются внутренние функции эпидермальных клеток, они начинают продуцировать цитокины и медиаторы, которые запускают механизм воспаления, усиливают пролиферацию клеток поражённой кожи. Так появляются доброкачественные лимфоидные образования дермы.
Экспрессия CD30-антигена атипичными лимфоидными клетками в сочетании с возможностью спонтанного перерождения лимфоматоидного папулеза в злокачественную лимфому свидетельствуют в пользу точки зрения тех дерматологов, которые считают лимфоматоидный папулёз разновидностью опухоли с небольшой степенью злокачественности. С другой стороны, редкость подобной трансформации говорит в пользу теории лимфоплазии. Таким образом, на текущий момент единого мнения не существует. Точно известно лишь то, что если пролиферация лимфоцитов перестанет подчиняться контролю иммунной системы, разовьётся злокачественная лимфома. Это свидетельствует о тесной связи иммунных и онкологических процессов в организме пациента.
Классификация
В современной дерматологии лимфоматоидный папулез классифицируют по гистологической картине, которая помогает в постановке диагноза. Существуют четыре разновидности лимфоматоидного папулеза:
Тип А. В гистологической картине встречаются разные виды клеток — анапластические лимфоидные клетки, лимфоциты, эозинофилы, нейтрофилы, гистиоциты; процесс смешанноклеточный;
• Тип В. Напоминает грибовидный микоз, в гистологическом препарате присутствуют единичные атипичные анапластические клетки злокачественной лимфомы, встречается редко;
• Тип С. Гистологически схож с крупноклеточной лимфомой, в препарате преобладают атипичные лимфоидные клетки;
• Тип D. Напоминает педжетоидный ретикулёз, наиболее агрессивен из всех типов.
Дифференциальная диагностика
Дифференцируют заболевание с парапсориазом, лимфомами, крапивницей, медикаментозной токсидермией, чесоткой, синдромом Сезари, лимфогранулематозом.
Радикальных методов терапии лимфоматоидного папулеза нет. Применяют большие дозы тетрациклинов в сочетании с кортикостероидами, сульфонами (дапсон) и цитостатиками (кармустин). Иногда используют ретиноиды, интерфероны. При диссеминированных высыпаниях проводится облучение электронным пучком, ПУВА-терапия. Однако ни один из методов терапии не дает долгосрочного эффекта. Прогноз зависит от течения лимфоматоидного папулеза, количества рецидивов. Необходимо регулярное диспансерное наблюдение у дерматолога.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Дифференциальная диагностика
Названия
Название: L41,2 Лимфоматоидный папулез.
L41.2 Лимфоматоидный папулез
Описание
Лимфоматоидный папулез. Доброкачественное заболевание лимфатической системы с рецидивирующими полиморфными кожными проявлениями. Клинически сопровождается папулезными высыпаниями различной величины, которые затем трансформируются в буллы, вскрываются, образуя эрозии, корочки, рубцы. Сыпь может располагаться на любом участке кожного покрова, субъективные ощущения отсутствуют. Диагноз выставляется на основании анамнеза, клинических данных, гистологического и иммуногистохимического исследований. В рамках лечебного курса лимфоматоидного папулеза применяются глюкокортикостероиды, антибиотики, цитостатики, ПУВА-терапия, однако все меры дают лишь кратковременный эффект.
Дополнительные факты
Лимфоматоидный папулез – узелковая лимфоидная опухоль кожи доброкачественного характера, имеющая тенденцию к медленному росту. Может быть представлена как единичным образованием, так и множественными элементами. Заболевание не имеет гендерной окраски, сезонности, эндемичности, наследственной предрасположенности. Средний возраст манифестации патологии — 40 лет. Распространенность в популяции составляет 1-2 случая на 1 млн. Человек. У представителей негроидной расы лимфоматоидный папулез практически не встречается. В самостоятельную нозологическую единицу заболевание выделил В. Маколей в 1968 году, отсюда другое название лимфоматоидного папулеза – болезнь Маколея.
В настоящее время лимфоматоидный папулез принято относить к группе первичных CD30+ лимфопролиферативных патологий кожи, наряду с такими процессами, как псевдолимфома, лимфогранулематоз, атипичный регрессирующий гистиоцитоз, анапластическая крупноклеточная лимфома кожи. С перечисленными заболеваниями лимфоматоидный папулез сближает не только общность происхождения, но и нередкое одновременное или последовательное развитие нескольких патологий у одного пациента. Повышенная настороженность дерматологов и онкологов в отношении лимфоматоидного папулеза объясняется возможностью его трансформации в злокачественную Т-клеточную лимфому (в 10-20% случаев).
L41.2 Лимфоматоидный папулез
Причины
Этиология заболевания неизвестна. Патогенез также является предметом споров и обсуждений. Большинство дерматологов, основываясь на гистологической картине лимфоматоидного папулеза, которая выглядит как истинная лимфома, считают заболевание лимфоцитарной опухолью кожи низкой степени злокачественности. Другие рассматривают лимфоматоидный папулез в качестве ограниченной доброкачественной лимфоплазии, возникающей при участии CD4-лимфоцитов, которые регулируют силу иммунного ответа. По всей вероятности, истина находится где-то посередине.
В развитии лимфоматоидного папулеза играет роль нарушение в дифференцировке Т-лимфоцитов, когда в результате латентно текущих аутоиммунных и системных заболеваний, врождённого или приобретённого иммунодефицита, ионизирующего излучения они начинают мутировать, провоцируя усиленную пролиферацию эпидермальных клеток, как здоровых, так и генетически деструктированных. Параллельно изменяются внутренние функции эпидермальных клеток, они начинают продуцировать цитокины и медиаторы, которые запускают механизм воспаления, усиливают пролиферацию клеток поражённой кожи. Так появляются доброкачественные лимфоидные образования дермы.
Экспрессия CD30-антигена атипичными лимфоидными клетками в сочетании с возможностью спонтанного перерождения лимфоматоидного папулеза в злокачественную лимфому свидетельствуют в пользу точки зрения тех дерматологов, которые считают лимфоматоидный папулёз разновидностью опухоли с небольшой степенью злокачественности. С другой стороны, редкость подобной трансформации говорит в пользу теории лимфоплазии. Таким образом, на текущий момент единого мнения не существует. Точно известно лишь то, что если пролиферация лимфоцитов перестанет подчиняться контролю иммунной системы, разовьётся злокачественная лимфома. Это свидетельствует о тесной связи иммунных и онкологических процессов в организме пациента.
Классификация
В современной дерматологии лимфоматоидный папулез классифицируют по гистологической картине, которая помогает в постановке диагноза. Существуют четыре разновидности лимфоматоидного папулеза:
Тип А. В гистологической картине встречаются разные виды клеток — анапластические лимфоидные клетки, лимфоциты, эозинофилы, нейтрофилы, гистиоциты; процесс смешанноклеточный;
• Тип В. Напоминает грибовидный микоз, в гистологическом препарате присутствуют единичные атипичные анапластические клетки злокачественной лимфомы, встречается редко;
• Тип С. Гистологически схож с крупноклеточной лимфомой, в препарате преобладают атипичные лимфоидные клетки;
• Тип D. Напоминает педжетоидный ретикулёз, наиболее агрессивен из всех типов.
Дифференциальная диагностика
Дифференцируют заболевание с парапсориазом, лимфомами, крапивницей, медикаментозной токсидермией, чесоткой, синдромом Сезари, лимфогранулематозом.
Радикальных методов терапии лимфоматоидного папулеза нет. Применяют большие дозы тетрациклинов в сочетании с кортикостероидами, сульфонами (дапсон) и цитостатиками (кармустин). Иногда используют ретиноиды, интерфероны. При диссеминированных высыпаниях проводится облучение электронным пучком, ПУВА-терапия. Однако ни один из методов терапии не дает долгосрочного эффекта. Прогноз зависит от течения лимфоматоидного папулеза, количества рецидивов. Необходимо регулярное диспансерное наблюдение у дерматолога.
Источник