Лечение синдрома бартера в германии

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 3 июля 2019;
проверки требует 1 правка.
Синдро́м Ба́рттера (но́рмотензи́вный ги́перальдостерони́зм, неонатальный синдром Барттера) — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленной внешними (вторичными) нарушениями передачи сигнала ангиотензина II[2].
История[править | править код]
Синдром назван в честь доктора Фредерика Барттера, который вместе с доктором Pacita Pronove первым описал его в 1960 году и нескольких пациентов в 1962 году[2][3][4][5][5].
Характеристика синдромов Барттера и Гительмана[править | править код]
Синдромы характеризуется гипокалиемией, нормальным или пониженным артериальным давлением, и наличием гипохлоремического метаболического алкалоза[6].
Псевдо-синдром Барттера[править | править код]
Псевдо-синдром Барттера (Pseudo-Bartter’s syndrome) — симптомокомплекс, имитирующий проявления аналогичные синдрому Барттера, однако не сопровождается характерными генетическими дефектами и проявляется кистозным фиброзом[7], а также развивается на фоне злоупотребления слабительными средствами, диуретиками, намеренного вызывания рвоты[8][9]
Этиология[править | править код]
Тип наследования аутосомно-рецессивный, андротропизм[2]. Синдром вызывают мутации генов, кодирующих белки, обеспечивающие транспорт ионов в почечных канальцах[10].
Синдром может быть разделён на различные подтипы в зависимости от мутировавшего гена[11]:
| Название | Тип синдрома Барттера | Мутация гена, кодирующего | Дефект |
| Неонатальный синдром Барттера | тип 1 | NKCC2 | Na-K-2Cl symporter |
| Неонатальный синдром Барттера | тип 2 | ROMK | K+ каналы толстого восходящего колена петли Генле |
| Классический синдром Барттера | тип 3 | CLCNKB | Cl- каналы |
| Синдром Барттера с нейросенсорной тугоухостью | тип 4 | BSND[12] | Cl- channel accessory subunit |
| Синдром Барттера, ассоциированный с аутосомно-доминантной гипокальциемией | тип 5 | CASR[13] | activating mutation of the calcium-sensing receptor |
| Синдром Гительмана | — | SLC12A3 (NCCT) | Sodium-chloride symporter |
Патогенез[править | править код]
Первичное звено — потеря способности почек задерживать калий. Возможные причины: нарушение функции почечных канальцев, нарушение реабсорбции хлоридов в петле Генле (приводит к увеличению поступления натрия и воды в дистальные отделы канальца, где происходит секреция калия), гипомагниемия. Потеря калия ведёт к многочисленным метаболическим и гормональным расстройствам: нарушению транспорта веществ через клеточные мембраны, усилению продукции сосудорасширяющих простагландинов, снижение секреции альдостерона, повышение продукции простагландинов в почках[2].
Клиническая картина[править | править код]
Первые признаки заболевания обычно проявляются в течение первого года жизни. Клинические проявления синдрома[2]:
- адинамия,
- головная боль,
- полиурия,
- полидипсия (жажда),
- рвота,
- гипернатриемия,
- гипокалиемия,
- гиперальдостеронурия,
- алкалоз.
Диагностика[править | править код]
Пренатальная диагностика — синдром Барттера можно заподозрить при многоводии[14].
Лечение[править | править код]
Чувствительность к ангиотензину II повышается или полностью восстанавливается после нормализации объёма внеклеточной жидкости и при лечении ингибиторами простагландинсинтетазы (даже на фоне сохраняющейся гипокалиемии)[2].
Пациентам рекомендуют диету, обогащённую калием и приём спиронолактона для уменьшения потери калия с мочой.[15]
См. также[править | править код]
- Тубулопатии
- Синдром Фанкони
- Альдостерон
- Ангиотензин II
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ 1 2 3 4 5 6 Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 31. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- ↑ Bartter F.C., Pronove P., Gill JR Jr, MacCardle R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome (англ.) // Am J Med (англ.)русск. : journal. — 1962. — Vol. 33, no. 6. — P. 811—828. — doi:10.1016/0002-9343(62)90214-0. — PMID 13969763. Reproduced in Bartter F.C., Pronove P., Gill J.R., MacCardle R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. 1962 (англ.) // J. Am. Soc. Nephrol. (англ.)русск. : journal. — 1998. — Vol. 9, no. 3. — P. 516—528. — PMID 9513916.
- ↑ Proesmans W. Threading through the mizmaze of Bartter syndrome (неопр.) // Pediatr. Nephrol.. — 2006. — Т. 21, № 7. — С. 896—902. — doi:10.1007/s00467-006-0113-7. — PMID 16773399.
- ↑ 1 2 Whonamedit — Bartter’s syndrome
- ↑ Gitelman H.J., Graham J.B., Welt L.G. A new familial disorder characterized by hypokalemia and hypomagnesemia (англ.) // Trans Assoc Am Physicians (англ.)русск. : journal. — 1966. — Vol. 79. — P. 221—235. — PMID 5929460.
- ↑ Royal Brompton & Harefield Hospital > Pseudo-Bartter’s syndrome Retrieved Mars, 2011
- ↑ Metyas, Samy; Rouman, Heba; Arkfeld, Daniel G. Pregnancy in a Patient With Gouty Arthritis Secondary to Pseudo-Bartter Syndrome // JCR: Journal of Clinical Rheumatology. — Т. 16, вып. 5. — С. 219—220. — doi:10.1097/RHU.0b013e3181e9312a.
- ↑ Kinns, H; Housley, D; Freedman, D. B. Munchausen syndrome and factitious disorder: the role of the laboratory in its detection and diagnosis (англ.) // Annals of Clinical Biochemistry (англ.)русск. : journal. — 2013. — May (vol. 50, no. Pt 3). — P. 194—203. — doi:10.1177/0004563212473280. — PMID 23592802.
- ↑ Rodriguez-Soriano J. Bartter and related syndromes: the puzzle is almost solved (англ.) // Pediatr Nephrol : journal. — 1998. — Vol. 12, no. 4. — P. 315—327. — doi:10.1007/s004670050461. — PMID 9655365.
- ↑ Naesens M., Steels P., Verberckmoes R., Vanrenterghem Y., Kuypers D. Bartter’s and Gitelman’s syndromes: from gene to clinic (англ.) // Nephron Physiol : journal. — 2004. — Vol. 96, no. 3. — P. p65—78. — doi:10.1159/000076752. — PMID 15056980.
- ↑ Zaffanello M., Taranta A., Palma A., Bettinelli A., Marseglia G.L., Emma F. Type IV Bartter syndrome: report of two new cases (неопр.) // Pediatr. Nephrol.. — 2006. — Т. 21, № 6. — С. 766—770. — doi:10.1007/s00467-006-0090-x. — PMID 16583241.
- ↑ Vezzoli G., Arcidiacono T., Paloschi V., et al. Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome (англ.) // J. Nephrol. : journal. — 2006. — Vol. 19, no. 4. — P. 525—528. — PMID 17048213.
- ↑ Dane B., Yayla M., Dane C., Cetin A. Prenatal diagnosis of Bartter syndrome with biochemical examination of amniotic fluid: case report (англ.) // Fetal. Diagn. Ther. : journal. — 2007. — Vol. 22, no. 3. — P. 206—208. — doi:10.1159/000098719. — PMID 17228161.
- ↑ Bartter Syndrome: Tubular and Cystic Kidney Disorders: Merck Manual Home Edition. Дата обращения 31 декабря 2007. Архивировано 28 июля 2012 года.
Ссылки[править | править код]
Источник

Синдром Барттера – это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.
Общие сведения
Синдром Барттера в клинической урологии представляет собой редкую генную мутацию — дефект петли Генле, наследуемую по аутосомно-рецессивному типу и проявляющуюся, как правило, уже в детском возрасте. Неспособность почечных нефронов задерживать калий приводит к хронической потере его с мочой и уменьшению объема циркулирующей крови при нормальном или пониженном АД. В зависимости от вида пораженных генов различают: неонатальный синдром Барттера 1 и 2 типов, классический синдром Барттера, синдром Гительмана. Также встречается приобретенный синдром псевдо-Барттера, характеризующийся сходными проявлениями, но не сопровождающийся патологией почечных канальцев.
Синдром Барттера
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагностика
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике — сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Источник
Синдром Бартера у детей. Диагностика и лечение
Врожденные нарушения транспорта в почечных канальцах формируют спектр редких состояний, при каждом из которых поражаются определенные сегменты нефрона. Успехи генетики и молекулярной биологии позволили расшифровать патогенез многих таких заболеваний и углубили наши представления о регуляции водно-электролитного обмена в норме.
Синдром Бартера редкая форма гипокалиемического метаболического алкалоза с гиперкальциурией наследуется аутосомно-рецессивно. Различают два клинических подтипа синдрома Бартера. Антенатальный синдром Бартера (называемый также синдромом гиперпродукции простагландина Е) обычно проявляется у новорожденных и протекает тяжелее, чем классический синдром Бартера; он включает многоводие в анамнезе, потерю соли и выраженное обезвоживание.
Более легкий классический фенотип проявляется позднее задержкой развития ребенка и частыми эпизодами обезвоживания в анамнезе. Фенотипически сходный синдром Гительмана обусловлен другим генетическим дефектом. Описан также вариант антенатального синдрома Бартера с нейросенсорной глухотой и ХПН, имеющий другую генетическую основу.
Патогенез синдрома Бартера у детей. Биохимические сдвиги при синдроме Бартера (гипокалиемический метаболический алкалоз с гиперкальциурией) напоминают последствия применения петлевых диуретиков и отражают нарушение транспорта натрия, хлорида и калия в восходящем отделе петли Генле. Потеря натрия и хлорида, приводящая к уменьшению внутрисосудистого объема, стимулирует ренин-ангиотензин-альдостероновую систему.
Альдостерон усиливает реабсорбцию натрия и секрецию калия, тем самым усугубляя гипокалиемию. Он усиливает также секрецию ионов водорода в дистальных отделах нефрона, что усугубляет метаболический алкалоз. Гипокалиемия стимулирует синтез простагландинов, которые еще больше активируют ренин-ангиотензин-альдостероновую систему. В основе синдрома Бартера лежат три разных генетических дефекта транспортеров, функционирующих на уровне петли Генле.
Каждый из них тем или иным образом участвует в транспорте натрия и хлорида. При антенатальном синдроме Бартера обнаруживаются мутации гена, кодирующего натрий/калий/2 хлоридный транспортер NKCC2 (объект действия фуросемида), или гена люминальных калиевых каналов (ROMK), тогда как для классического синдрома характерны дефекты базолатеральных хлоридных каналов (ClC-Kb).
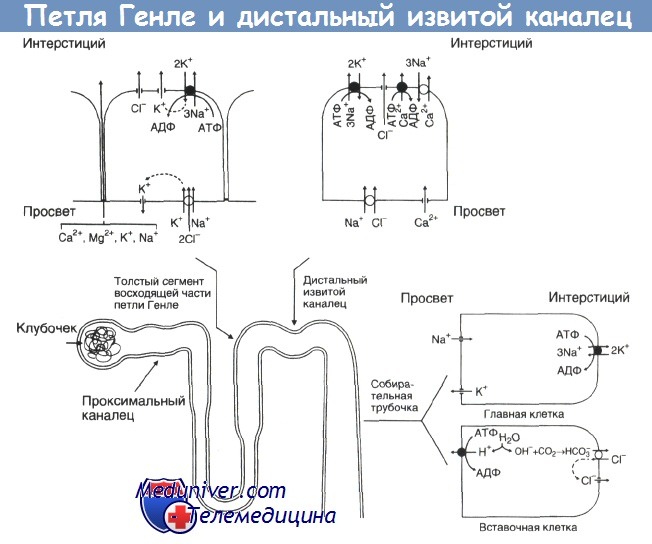
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Клинические проявления синдрома Бартера у детей. Как выясняется из анамнеза, при беременности было многоводие. У новорожденного возможна дизморфия, например треугольное лицо, оттопыренные уши, косоглазие и опущение углов рта. Родственная связь между родителями указывает на аутосомно-рецессивное наследование синдрома. Для более позднего возраста характерны повторные эпизоды обезвоживания, задержка развития и биохимические нарушения. При синдроме Бартера всегда имеют место гипокалиемия и метаболический алкалоз.
Содержание кальция в моче обычно повышено. Часто значительно повышен уровень ренина, альдостерона и простагландина Е в сыворотке крови, особенно при тяжелой антенатальной форме синдрома. АД в большинстве случаев нормальное, хотя обезвоживание при выраженной потере соли у больных с антенатальной формой синдрома может приводить к артериальной гипотонии. Функция почек, как правило, сохранена. При УЗИ иногда обнаруживается нефрокальциноз как следствие гиперкальциурии.
Диагностика синдрома Бартера у детей. Диагноз устанавливают на основании клинической картины и лабораторных данных. У новорожденных о синдроме Бартера свидетельствует гипокалиемия (обычно ниже 2,5 ммоль/л) на фоне метаболического алкалоза. В типичных случаях отмечается гиперкальциурия. Гипомагниемию обнаруживают лишь у немногих больных; она более характерна для синдрома Гительмана. Поскольку проявления напоминают последствия продолжительного использования петлевых диуретиков, всегда необходимо выяснить, применялись ли эти средства (даже у маленьких детей).
Аналогичная клиническая картина имеет место при хронической рвоте, но при этом содержание хлорида в моче снижено, тогда как при синдроме Бартера оно повышено. При гистологическом исследовании почек находят гиперплазию юкстагломерулярного аппарата. Однако диагностическая биопсия при синдроме Бартера выполняется редко.
Лечение и прогноз синдрома Бартера у детей. Терапия синдрома Бартнера направлена на предотвращение обезвоживания и поддержание питания и коррекцию гипокалиемии. Зачастую требуются очень большие дозы калия, но и в этих случаях его уровень в сыворотке не всегда удается нормализовать, особенно у новорожденных. Грудные и маленькие дети могут нуждаться и в добавках натрия. Эффективен также индометацин, ингибирующий синтез простагландинов.
При внимательном отношении к электролитному балансу, объемному статусу и росту ребенка долговременный прогноз обычно благоприятный. Однако хроническая гипокалиемия, нефрокальциноз, длительное введение индометацина могут иногда приводить к развитию интерстициального нефрита и ХПН.
— Также рекомендуем «Синдром Гительмана у детей. Диагностика и лечение»
Оглавление темы «Почечная недостаточность у детей»:
- Рахит при канальцевом ацидозе. Нефрогенный несахарный диабет у детей
- Диагностика нефрогенного несахарного диабета у детей. Лечение
- Синдром Бартера у детей. Диагностика и лечение
- Синдром Гительмана у детей. Диагностика и лечение
- Острый тубулоинтерстициальный нефрит у детей. Диагностика и лечение
- Хронический тубулоинтерстициальный нефрит у детей. Диагностика и лечение
- Некроз коркового вещества почек у детей. Диагностика и лечение
- Острая почечная недостаточность у детей. Причины
- Клиника острой почечной недостаточности у детей. Диагностика
- Лечение острой почечной недостаточности у детей. Инфузии
Источник