Кофейное пятно код по мкб

Пятна округлой или овальной формы с четкими границами размером от 1 до 20 см и более (в среднем 2-5 см у взрослых),равномерной светло- или темно-коричневой окраски,напоминающие цвет «кофе с молоком» или загар.Локализация любая,но чаще на туловище,ягодицах и нижних конечностях,описаны случаи появления пятен в области гениталий и слизистых облочек рта.Пятна существуют в течении всей жизни,могут увеличиваться в размерах до полового созревания,после которого процесс стабилизируется, а в некоторых
случаях происходит сокращение диаметра.Количество пятен не более трех считается нормальным
состоянием.О развитии меланомы и других злокачественных опухолей кожи в местах локализации пятен не сообщалось.При количестве пятен более трех,»изрезанности» их границ,блашкоидном и(или) сегментарном расположении необходимо исключать некоторые генодерматозы.
(Multiple familial café au lait spots)
(Mc-Cune-Albright syndrome)
Патология костей.
- Полиостотическая фиброзная дисплазия — замещение костного вещества соединительной тканью, что приводит к деформации костей и патологическим переломам.
- Очаги поражения костей часто расположены ипсилатерально по отношению к пятнам цвета кофе с молоком.
Эндокринные нарушения: преждевременное половое созревание (преимущественно у девочек), гипертиреоз, синдром Кушинга
(Silver-Russell-syndrome)
(Bloom syndrome)
(Watson syndrome)
(Westerhof syndrome)
(Leschke’s syndrome)
(Tuberous sclerosis)
(Noonan syndrome)
(Legius syndrome)
(Fanconi anemia)
multiple Lentigines, Electrocardiographic conduction abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth,sensorineural Deafness
Наследуется по аутосомно-доминантному типу. Характеризуется генерализованными рассеянными по всему кожному покрову (за исключением лица) лентигинозными высыпаниями, которые обычно сочетаются с различными аномалиями сердца (гипертрофия, отклонение оси сердца, стеноз легочной артерии и др.), а также аномалиями половых органов, нейросенсорной глухотой и др.
(Capute-Rimoin-Konigsmark Syndrome)
с олигофренией,
врожденной глухотой и умеренно выраженным дизрафическим статусом в виде частичной синдактилии,расщепления задних отростков нижних грудных и верхних поясничных позвонков
(Jaffe-Campanacci syndrome)
(Cardio-Facio-Cutaneous Syndrome)
Грубые изменения лица, глаз, ушей, шеи, скелета, грудной клетки, микрогнатия, лимфатические отеки, низкий рост, потеря слуха,ихтиозоподобный гиперкератоз, гемангиомы, множественные пятна цвета кофе с молоком, многочисленные складки на ладонях и стопах, редкие, грубые ломкие волосы, выпадают ресницы, брови, тонкие блестящие ногти. Череп большой, удлиненный и сдавленный с боков. Часто тяжелое поражение психики, слабоумие, выраженные поражения желудочно-кишечного тракта, подслизистая волчья пасть, высокое, суженное
небо, недоразвитие супраорбитальных дуг
(Ataxia telangiectasia)
Развитие мозжечковой атаксии и телеангиэктазий. Первично телеангиэктазии чаще локализуются на конъюнктиве глаз. Сосудистые поражения появляются также на шее, верхней части груди, лице и на ушных раковинах. Поздние кожные проявления включают в себя участки гипо- или гиперпигментации, пятна цвета кофе с молоком, гирсутизм, генерализованный экзематозный дерматит, гранулематозные поражения кожи. Иногда наблюдают преждевременное поседение волос
(Constitutional MMR deficiency syndrome)
(Gaucher disease)
- безболезненная гепатомегалия и спленомегалия .
- гиперспленизм и панцитопения
- неврологические симптомы,болезнь Паркинсона
- остеопороз у 75% пациентов
- пятна цвета кофе с молоком
(Carney syndrome)
(Tay syndrome 1)
(Johnson-McMillin syndrome)
(Fevre-Languepin syndrome)
(Basal cell nevus syndrome)
Множественные невоидные элементы,мелкие фибромы,комедоны,бородавки, веснушки,пятна цвета кофе с молоком, аномалии зубов,глаз,деформации ребер, фибромы и кисты яичников, лимфатические мезентериальные кисты, spina bifida, заячья губа, волчья пасть, кальцификация твердой мозговой оболочки, множественные кисты трубчатых костей, мозговые аномалии, липомы, гипогенитализм, аномалии лицевого черепа, снижение интеллекта.
(Mukamel syndrome)
(Gastro-cutaneous syndrome)
(Рartial unilateral lentiginosis)
(Multiple endocrine neoplasia 2b)
Источник

Библиотека
Premium Aesthetics
БЕСПЛАТНАЯ КОНСУЛЬТАЦИЯ: поможем врачам и владельцам клиник выбрать оборудование для лечения кофейных пятен
Кофейные пятна (пятна цвета кофе с молоком, café au lait spots, café au lait macules, CALMs) — это гиперпигментированные участки на коже с неровными или сглаженными краями, цвет которых варьируется по от светло- до темно-коричневого. В зарубежной литературе их часто называют «café au lait spots», что является сочетанием французского «café au lait» (кофе с молоком) и английского «spots» (пятна).
В нашей компании Вы можете приобрести следующее оборудование для удаления кофейных пятен:
- M22 (Lumenis)
У новорожденных кофейные пятна возникают с различной вероятностью, в зависимости от фототипа кожи:
- 0,3% детей — I–III фототипы;
- 3% детей — IV фототип;
- 18% детей — V–VI фототипы.
До наступления пубертата такие пятна могут появиться с вероятностью 13% у светлокожего населения и 27% — у темнокожего.
Этиология и патогенез
Кофейные пятна обычно говорят о наличии у пациента нейрофиброматоза I типа — мультисистемного заболевания, для которого характерны пятна на спине, шее, в подмышечных областях, а также скелетные дисплазии, доброкачественные или злокачественные опухоли из оболочек нервов (нейрофибромы). Средняя частота появления нейрофиброматоза I типа составляет 1:3000. Его основными причинами считаются мутации или делеции (потери участка) гена NF1. Этот ген несет ответственность за продукцию нейрофибромина 1 — белка, который подавляет возникновение опухолей нервной системы. Снижение его синтеза ведет к появлению нейрофибром и других клинических признаков заболевания.
Кофейные пятна на коже также могут возникать по следующим причинам:
- Синдром Олбрайта-МакКьюна-Штернберга — преждевременное половое созревание, множественная фиброзная остеодисплазия, гиперпигментация кожи.
- Болезнь Бурневилля-Прингла — туберкулезный склероз; редкое генетическое заболевание, при котором в органах и тканях образуются множественные доброкачественные опухоли.
- Анемия Фанкони — проявляется комплексом симптомов, наиболее серьезными из которых являются гематологические нарушения и опухоли.
- Синдром Коффина-Сириса — характеризуется аплазией или гипоплазией дистальной фаланги или ногтя на V пальцах стоп, задержкой в развитии, умственной отсталостью, грубыми чертами лица и др.
Пятна на коже цвета кофе с молоком могут быть следствием RAS-мутаций — изменений в семействе генов и белков RAS. Это малые ГТФазы (ферменты, связывающие и гидролизующие гуанозинтрифосфат — энергетический субстрат для РНК), которые участвуют в передаче сигнала извне клетки и регулируют клеточное деление. Некоторые мутации RAS способствуют появлению и метастазированию опухолей.
У 50% пациентов генетические мутации, ответственные за возникновение кофейных пятен на коже, происходят спонтанно. То есть наследственный механизм передачи заболевания в данном случае имеет опосредованное значение.
Что касается патогенеза, то кофейные пятна появляются на фоне роста продукции меланина и наличия в коже гигантских меланосом. У пациентов с нейрофиброматозом I типа число меланоцитов в пигментных пятнах увеличивается более значительно по сравнению с другими заболеваниями.
Клинические проявления
Диагностика нейрофиброматоза I типа строится на поиске ключевых критериев — о заболевании можно говорить при наличии как минимум 2 из 7 признаков:
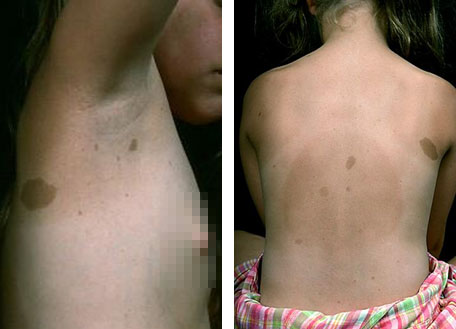
- Шесть и более кофейных пятен или макул на коже диаметром более 5 мм у детей (до пубертата) и более 15 мм у подростков и взрослых (после пубертата) (рис. 1).
- Более двух пятен в паху или в подмышечных областях.
- Две и более типичные нейрофибромы или одна плексиформная нейрофиброма.
- Глиома зрительного нерва.
- Две и более гамартомы радужки глаза — обычно выявляются офтальмологом с помощью щелевой лампы.
- Дисплазия клиновидной кости черепа или патологии длинных костей скелета (например, псевдоартроз).
- Семейный анамнез — наличие нейрофиброматоза у ближайших родственников (отца или матери, братьев или сестер).
Многие из этих признаков отсутствуют до наступления пубертата, что затрудняет диагностику у детей. При этом если пациент из группы риска достигает 10-летнего возраста без появления хотя бы одного симптома (например, кофейных пятен), в дальнейшем он почти наверняка не будет иметь нейрофиброматоза I типа.
Нейрофиброматоз I типа следует отличать от II типа, при котором кожные проявления сравнительно бедные, зато у пациентов имеется склонность к возникновению менингеом и билатеральных акустических неврином.
Другие синдромы, при которых на коже могут возникать кофейные пятна (часть из них была указана выше):
- Синдром Сильвера-Расселла — задержка роста и психомоторного развития, треугольное лицо, укорочение пальцев рук, синдактилия, характерные светло-коричневые пятна на коже и др.
- Синдром Блума — карликовость, сниженный иммунитет, повышенная фоточувствительность кожи, кофейные пятна и др.
- Синдром Горлина-Гольца — наследственная базальноклеточная карцинома кожи с эритематозными поражениями и аномалиями скелета.
- Синдром Маффуччи — доброкачественное разрастание хрящевой ткани с деформацией костей и темными сосудистыми образованиями на коже.
Это далеко не все синдромы, при которых на коже может возникать характерная гиперпигментация цвета кофе с молоком. Однако их объединяет два важных фактора — относительная редкость и общая тяжесть, т.е. наличие других признаков (кроме кофейных пятен), оказывающих гораздо более значимое влияние на организм.
В целом отмечены следующие закономерности:
- Множественные кофейные пятна небольшого размера говорят о наличии одного из вышеназванных заболеваний (обычно это нейрофиброматоз I типа).
- Единичные крупные светло-коричневые пятна, как правило, не связаны с другими патологиями и существуют сами по себе.
Рис. 1. Кофейные пятна в подмышечной области и на спине (www.medscape.com)
Принципы лечения
До начала терапии кофейных пятен следует исключить более серьезные патологии. Для этого пациента необходимо внимательно опросить, тщательно осмотреть, назначить лабораторные и инструментальные исследования. Сделать это в косметологическом кабинете невозможно, поэтому важно соблюдать преемственность и поддерживать связь с врачами других специальностей.
Если кофейные пятна ассоциированы с какими-либо заболеваниями, следует направить максимум усилий на лечение основной патологии. Если же кофейные пятна существуют сами по себе, или основное заболевание уже пролечено, а участки гиперпигментации остались, можно подключить аппаратные методы.
Одной из таких методик является Q-switched александритовый лазер. В исследовании с участием 48 человек он показал хорошую и отличную эффективность у более половины пациентов (51,4%) после в среднем 3,2 сеансов. При этом рецидив зафиксирован в 10,4% случаев. Лазерное лечение кофейных пятен несет в себе высокий риск поствоспалительной гиперпигментации — около 50%. Если она произошла, следует прекратить терапию до тех пор, пока гиперпигментация не пройдет.
Для лечения кофейных пятен также с успехом применяется IPL-терапия. Интенсивный импульсный свет (IPL) избирательно разрушает меланин в участках его избыточного накопления, что приводит к осветлению кожи. Современные IPL-аппараты, такие как M22 (Lumenis), снабжаются светофильтрами, которые позволяют воздействовать на кожу необходимым диапазоном спектра излучения. Благодаря этому, IPL можно безопасно применять для лечения кофейных пятен на темной коже — вплоть до V фототипа по Фитцпатрику.
Замечено, что если кофейное пятно удалено полностью, то риск его повторного появления минимален. Если же пятно удалено частично, вероятность рецидива составляет 50%.
В нашей компании вы можете приобрести самое современное косметологическое оборудование
- Аппараты для фотоомоложения
- Аппараты с IPL (Intensive Pulse Light)
- Многомодульные косметологические платформы
Аппараты — Кофейные пятна
M22
Многомодульная платформа IPL, фракционный лазер, Nd:YAG, Nd:YAG QS
FotoFinder aesthetics
Диагностические комплексы для диагностики в эстетической медицине, фотодокументирования, маркетинга услуг
Все Аппараты
Видео — Кофейные пятна
Все Видео
Другие Показания
Источник
Общие сведения
Нейрофиброматоз — это группа наследственных относительно редких заболеваний, проявляющихся множеством симптомов и фенотипической вариабельностью. Наиболее часто нейрофиброматоз (НФ) проявляется характерными патологическими изменениями — развитием опухолей преимущественно эктодермального происхождения с поражением кожного покрова, центральной нервной системы, наличием специфических пигментных пятен на коже по цвету «кофе с молоком», аномалиями развития костного скелета и рядом других проявлений.
Код нейрофиброматоз по мкб-10: Q85.0 (нейрофиброматоз незлокачественный). Нейрофиброматоз относится к наиболее распространенной форме наследственной моногенной патологии, частота встречаемости которого варьирует в пределах 1:2000 — 1:40000. Характерен аутосомно-доминантный тип наследования заболевания с частотой проявления (пенетрантностью) приближающейся к 100%. Различий в частоте поражения по половому признаку не выявлено.
В целом по клинико-морфологическим проявлениям выделено и описано восемь типов нейрофиброматоза, наиболее значимыми из которых являются:
- Нейрофиброматоз первого типа (НФ 1). Около 90% всех нейрофиброматозов приходится на тип НФ1, который и является наиболее распространённым среди заболеваний этой группы.
- Нейрофиброматоз второго типа (НФ 2). Встречается значительно реже (1:40000), но имеет более агрессивное течение, чем НФ1.
Некоторые авторы считают, что из восьми форм нейрофиброматоза все, кроме НФ2 считаются абортивными и не должны выделяться в качестве самостоятельных нозологических форм.
Нейрофиброматоз 1 типа (синоним «болезнь Реклингаузена»)
Частота встречаемости у новорожденных составляет 1:4000, в русской популяции — 1,28:10 000. Более характерна передача по отцовской линии, чем по материнской. В более половине случаев проявления заболевания минимальны, часто встречаются неполные и моносимптомные формы. Около 80% случаев нейрофиброматоза спорадические и являются результатом новых мутаций. В связи с присущей гену NF1 высокой частоты мутаций, определяющих клиническое развитие нейрофиброматоза, предполагается или чрезвычайно высокая его мутабельность или наличие мутаций в нескольких локусах гена.
Для нейрофиброматоза I типа характерен выраженный полиморфизм клинических проявлений, прогрессирующее течение и частое вовлечение в процесс различных органов и систем с развитием тяжелых осложнений, нередко приводящих к летальному исходу. Болезнь Реклингхаузена (нейрофиброматоз) имеет четко прослеживающуюся тенденцию к медленному прогрессированию. Существует два периода резкого увеличения активности патологического процесса: первый от 5 и до 10 и второй от 35 до 50 лет. При этом, второй период активности в подавляющем числе случаев (около 75%) обусловлен малигнизацией опухолевых новообразований. К провоцирующим факторам, способствующим росту нейрофибром, относятся состояния и возрастные изменения, сопровождающиеся гормональной перестройкой организма: беременность, период полового созревания.
Особенность заболевания является специфическая последовательность манифестации симптоматики нейрофиброматоза 1-го типа в зависимости от возраста пациента, что и обуславливает трудности в его диагностике у детей раннего возраста. Так, нейрофиброматоз у детей первых лет жизни/с рождения может присутствовать только в виде некоторых признаков заболевания, чаще — это крупные пигментные пятна или скелетные дисплазии и плексиформные нейрофибромы, а другие характерные симптомы могут манифестировать значительно позже (к 5-15 годам). А если учесть, что степень их выраженности, течение и скорость прогрессирования патологического процесса варьируют в широких пределах становится понятными затруднения в постановке диагноза, что и способствует его поздней диагностике.
Нейрофиброматоз 2 типа
Нейрофиброматоз II типа встречается независимо от НФ-I типа; его проявления определяются локализацией основного патологического процесса и соответствует симптоматике поражения головного/спинного мозга. В спинном мозге чаще в процесс вовлекаются оболочки спинальных корешков шейного и поясничного отделов в виде Шванном (опухоль доброкачественная из шванновских клеток нервных оболочек), конский хвост (задние корешки); при локализации в головном мозге в процесс могут вовлекаться черепно-мозговые нервы, продолговатый мозг и Варолиев мост. Реже в процесс вовлекаются периферические нервы с развитием эпендимом и менингиом.
Средний возраст манифестации признаков заболевания при НФ 2 составляет 20 лет, а на момент постановки диагноза – около 28 лет. Как правило, НФ 1 начинает проявляться в раннем детстве, преимущественно с кожных симптомов, в то время как НФ 2 проявляется в молодом возрасте, преимущественно с развития глухоты, обусловленной развитием вестибулярных шванном и других симптомов, вторичных относительно спинальных шванном и менингиом.
Патогенез
В основе патогенетических изменений нейрофиброматоза 1 типа лежит высокая частота спонтанных мутаций гена NF1 (17q11.2), кодирующего цитоплазматический белок-онкосупрессор нейрофибромин, который вырабатывается в нервных и специализированных клетках нейроглии. Нейрофибромин содержит в своем составе особый домен, который и реализует супрессорный эффект относительно пролиферации клеток преимущественно нейроэктодермального происхождения за счет инактивации адаптерного белка Ras, который является ингибитором структуры митогенных сигнальных путей. При такого рода генетическом дефекте в хромосомах 17q происходит нарушение динамического равновесия регуляции роста и его смещение в сторону пролиферации и образования доброкачественного опухолевого роста.
Описано более 500 видов мутаций в гене на хромосоме 17q, которые и препятствуют осуществлению регулирующей функции гена NF1 в онкогенезе. Механизм развития клинических проявлений до настоящего времени точно неизвестен. Принято считать, что в основе патологии лежит развитие из нервных оболочек различных опухолей (невриномы, нейрофибромы). Примерно половина случаев являются следствием новых мутаций.
Генетический дефект при нейрофиброматозе 2 типа располагается в 22 хромосоме (22q12) и непосредственно кодирует процесс синтеза белка Мерлина, также являющегося супрессором опухолевого роста. Наибольшее значение он имеет в регулировании пролиферации (размножения) клеток нейроэктодермального происхождения. При мутации синтеза Мерлина в одной хромосоме не проявляется на клеточном уровне, а при симметричной мутации (повреждении) аллельного гена в клетке прекращается синтез Мерлина и соответственно, присущее здоровому организму равновесие в регуляции роста смещается (дрейфует) в сторону пролиферации с развитием процесса доброкачественного опухолевого роста.
Классификация
В основе классификации нейрофиброматоза лежат клинико-морфологические особенности заболевания в соответствии с чем выделяют:
- Периферический нейрофиброматоз (1 тип)— в клинической картине доминируют поражение периферических нервов и кожных покровов.
- Центральный нейрофиброматоз (2 тип) — в патологический процесс вовлекаются преимущественно черепные нервы и спинальные корешки.
Причины
Этиологическим фактором нейрофиброматоза 1 типа являются множественные мутации одного из самых протяженных и сложно организованных генов NF1 (17q11.2), являющихся следствием инактивации преимущественно отцовской аллели. Причиной нейрофиброматоза 2 типа является генетический дефект в 22 хромосоме (22q12).
Симптомы
Нейрофиброматоз Реклингхаузена начинает проявляться в большинстве случаев множественными нейрофибромами, с локализацией по ходу периферических нервов, в виде округлых болезненных узелков, расположенных в толще кожи, различающиеся по своим размерам. Нейрофибромы представлены в виде узелков на/в толще кожи нормальной или синюшно-красной окраски и мягко эластической консистенции.
Характерным признаком, начиная с периода новорожденности/первых лет жизни детей является появление на коже мелких «кофейных пятен», напоминающих веснушки с четкими границами и ровными краями с преимущественной их локализацией на участках тела со складками (в паховой области, подмышечных впадинах, на животе). По мере взросления ребенка наблюдается тенденция к увеличению их численности (от единичных пятен до нескольких сотен) и размеров (от точечных до 2-5 сантиметров в диаметре), они также могут приобретать темную окраску. Следует учитывать, что к диагностически значимым пятнам относятся у детей до пубертата — диаметром более 5 мм и после полового созревания — 15 мм и более. Нейрофиброматоз (фото начальной стадии) приведены ниже.

Мягкие кожные опухоли, как правило, у детей отсутствуют, особенно первых лет жизни и возникают к 10-15 годам. При этом, их количество особенно возрастает в пубертатном периоде. При пальпации они безболезненны, однако при вовлечении в патологический процесс периферических нервов, могут возникают боли, гипестезии. На коже могут присутствовать и другие изменения: участки депигментации, сосудистые пятна, очаговое поседение волос, гипертрихоз. Возможно развитие нейрофибром. Плексиформная нейрофиброма может образовывать гигантские опухоли, состоящие из кожных и подкожных элементов.
Некоторые авторы (В.В. Мордовцева) выделяет четыре варианта клинического течения нейрофиброматоза I типа:
- с наличием незначительных пигментных пятен типа веснушек в сочетании с крупными «кофейными» пятнами при отсутствии нейрофибром или с наличием единичных опухолей;
- с наличием преимущественно крупных пигментных кофейных пятен на коже и небольшим количеством нейрофибром;
- с преобладанием нейрофибром и незначительным количеством пигментных пятен (по типу веснушек/крупных);
- смешанный тип.
Факультативные симптомы нейрофиброматоза проявляются изменениями в костной системе в виде различных деформаций позвоночного столба — сколиозы, лордосколиозы, кифосколиозы, псевдоартрозы, утончение/искривление длинных трубчатых костей, патологических переломов. Нередко развиваются лицевые дисморфии: аномалии глазных щелей, глазной гипертелоризм, деформация ушных раковин, неправильная форма черепа и др. Для пациентов с НФ-I характерны макроцефалия, низкий рост, дисплазия клиновидной кости и затылочной части черепа.
Характерными поражениями нервной системы являются судорожный и гипертензионно-гидроцефальный синдромы, опухоли ЦНС (эпендимома, астроцитома, глиома зрительного нерва/ствола мозга), эпилепсия, что появляется соответствующей симптоматикой. Так, глиома зрительного нерва проявляется нарушением зрения.
Для NF1 характерны дополнительные проявления в виде когнитивных расстройств от легких до выраженных, часто в сочетании с умеренным снижением IQ ребенка, затруднением в освоении чтения, письма, математики и нервно-психической неуравновешенностью; наличие эндокринных расстройств (полового созревания, феохромоцитома, нарушение роста и др.). Со стороны сосудов у пациентов часто регистрируется стеноз почечных артерий с повышением АД, окклюзия артерий, болезнь Мойа-Мойа.
Клиническая манифестация заболевания НФ2 включает развитие двусторонних вестибулярных шванном или шванном других черепных периферических и спинальных нервов при минимальных экстраневральных и кожных симптомах. Двусторонняя шваннома (невринома) слухового нерва относится к основным признакам, который встречается практически в 90% случаев, редко обнаруживается у детей, развиваются преимущественно в позднем подростковом/раннем взрослом возрасте. НФ-II.
Значительно реже (около 10%) случаев встречаются различного рода менингиомы (спинальные, интракраниальные, менингиомы зрительных нервов), глиомы и эпиндимомы. Первым симптомом часто является снижение/потеря слуха, появление в ушах шума, который может сопровождаться атаксией и головокружением. Пятна на коже цвета «кофе с молоком» в большинстве случаев отсутствуют или присутствуют в небольшом количестве. У 50-70% больных нейрофиброматозом 2 типа диагностируются зрительные нарушения (гемартромы, катаракты, ретинобластомы, менингиомы зрительных нервов).
Ниже в таблице приведены характерные проявления нейрофиброматоза 1 и 2 типа.

Анализы и диагностика
Диагноз нейрофиброматоза 1 типа ставится на основе абсолютных и относительных клинических признаков и данных инструментальных методов диагностики:
- КТ/МРТ позвоночника, головного мозга, внутренних органов.
- Рентгенография позвоночника.
- Электрокохлеография.
- Офтальмоскопия.
- Импедансометрия.
- Генетическое обследование и проведение генеалогического анализа.
Лечение
Патогенетическое лечение нейрофиброматоза до настоящего времени не разработано. В большинстве случаев проводится симптоматическая терапия, что и подтверждает специализированный форум. Так, при выраженном болевом синдроме показаны НПВС (Диклофенак, Мелоксикам, Ибупрофен, Нимесулид, Напроксен, Кетопрофен, Пироксикам), неопиодные (Парацетамол, Анальгин, Бутадион, Аспирин и др.) и опиоидные анальгетики (Трамадол, Фентанил, Налбуфин, Тримеперидин), трициклические антидепрессанты (с осторожностью из-за высокого риска судорожного синдрома), Нейронтин (Габапентин), Топирамат. Консервативная терапия предусматривает курсовое назначение препаратов, влияющие на:
- дегрануляцию тканевых базофилов (назначается Кетотифен);
- пролиферацию клеточных элементов (назначаются ретиноиды — Ацитретин, Этретинат, Бексаротен, Тазаротен);
- снижение во внеклеточном матриксе количества гликозаминогликанов (Гиалуронидаза).
Согласно последним рекомендациям, лечение нейрофиброматоза может проводиться с использованием новых перспективных препаратов для таргетной (нацеленной) терапии, воздействующие на определенное звено в патогенетической цепи нейрофиброматоза. Для таргетной терапии используются два вида молекул: мелкие молекулы и моноклональные антитела. К таким препаратам относятся Бевацизумаб (Авастин), Эрлотиниб, Иматиниб, Салиразиб, Лапатиниб (Тайверб).
В случаях малигнизации опухолей проводится химиотерапия и лучевая терапия. При когнитивных нарушениях и плохой обучаемости детей рекомендуется проводить учебный процесс в спецшколах, а также проведение социальной реабилитации взрослых пациентов.
Доктора
Лекарства
- Кетотифен, Этретинат, Тазаротен, Ацитретин, Гиалуронидаза, Диклофенак, Мелоксикам, Ибупрофен, Нимесулид, Напроксен, Кетопрофен, Пироксикам.
- Парацетамол, Анальгин, Бутадион, Аспирин, Трамадол, Фентанил, Налбуфин, Тримеперидин, Нейронтин (Габапентин), Топирамат, Бевацизумаб (Авастин).
- Эрлотиниб, Иматиниб, Салиразиб, Лапатиниб (Тайверб).
Процедуры и операции
При наличии сколиоза и костных деформаций показаны операции ортопедические. При наличии болезненных липом, нейрофибром