Код по мкб синдром прадера вилли

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Патогенез
- Симптомы
- Лечение
Другие названия и синонимы
Синдром гипотонии-ожирения.
Названия
Синдром Прадера — Вилли.
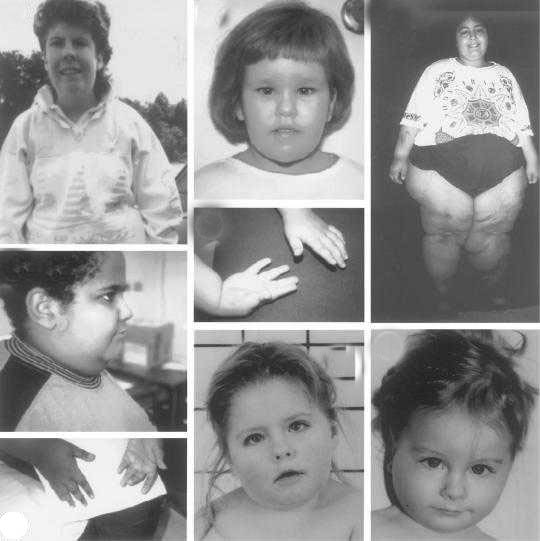
Проявления синдрома Прадера — Вилли
Синонимы диагноза
Синдром гипотонии-ожирения.
Описание
Синдром Прадера — Вилли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют или не экспрессируются примерно 7 генов из 15-й хромосомы, унаследованной от отца.
Кариотип 46 XX или ХУ, 15q-11-13. Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г.
По данным регистра ассоциации больных с синдромом Прадера-Вилли, в США и Канаде на декабрь 1986 г. Насчитывалось 1595 больных. В последние годы удалось установить популяционную частоту патологии, составляющую 1 : 10 000 — 1 : 20 000.
Причины
Авторы, впервые описавшие синдром, высказывали предположение об аутосомно-рецессивном типе наследования заболевания. Затем появились сообщения о возможности аутосомно-доминантной передачи болезни. Подтверждением данных гипотез могли служить наблюдавшиеся семейные случаи патологии. Однако большинство описанных клинических наблюдений синдрома Прадера — Вилли носило спорадический характер.
Последующие исследования позволили установить у детей с синдромом Прадера — Вилли определенные хромосомные нарушения. Цитогенетический анализ показал, что хромосомные аномалии у больных были представлены либо транслокациями (t 15/15), либо мозаицизмом. В 1987 г. Появились первые сообщения о микроделеции хромосомы 15. Однако окончательная идентификация хромосомных изменений при синдроме Прадера — Вилли стала возможной только после внедрения в практику молекулярно-генетических методов исследования.
В настоящее время установлено, что развитие синдрома Прадера — Вилли связано с повреждением критического района хромосомы 15 (сегмента q11,2- q13). При этом оказалось, что повреждение этого же участка хромосомы 15 наблюдается и при другом заболевании — синдроме Ангельмана, клиническая картина которого существенно отличается от синдрома Прадера — Вилли и характеризуется ранним (в возрасте 6-12 мес) замедлением психомоторного развития, микроцефалией, нарушением речи (в 100% случаев), атаксией, неконтролируемым насильственным смехом, частыми эпилептиформными припадками, специфическим выражением лица.
Таким образом, несмотря на повреждение при синдромах Прадера — Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
Объяснение фенотипических различий получено лишь в последние годы. Оказалось, что развитие этих заболеваний связано с новыми генетическими явлениями — геномным импринтингом и унипарентальной дисомией.
Геномный импринтинг — новое явление, открытое благодаря успехам молекулярной генетики. Он означает различную экспрессию генетического материала (гомологичных аллелей) в хромосомах в зависимости от отцовского или материнского происхождения, т. Е. Свидетельствует о влиянии родителей на фенотип ребенка. До настоящего времени считалось, что вклад в проявляемость (экспрессию) генов отца и матери равноценен.
По сути геномный импринтинг — это половой и тканевозависимый сложный модификатор генной активности некоторых локусов хромосом в зависимости от их родительского происхождения. Проявления геномного импринтинга выявлены и при других заболеваниях — синдромах Сотоса, Беквита-Видемана, Сильвера-Рассела, муковисцидозе и других.
Унипарентальная (однородительская) дисомия — наследование обеих хромосом только от одного из родителей. В течение многих лет считалось, что такое наследование невозможно. Лишь с помощью молекулярно-генетических маркеров удалось доказать возможность однородительской дисомии. Природа унипарентальной дисомии окончательно не выяснена, однако установлено, что она обязана своим происхождением ряду генетических и биохимических нарушений.
Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить микроделецию или унипарентальную дисомию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы — прометафазный анализ, использование ДНК-маркеров определенных участков хромосомы 15 (исследование процессов метилирования) и.
На сегодняшний день синдромы Прадера — Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений — геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера — Вилли может быть обусловлен двумя основными механизмами. Первый из них — микроделеция хромосомы 15 (15q11,2-q13), которая всегда отцовского происхождения. Второй — материнская изодисомия, т. Е. Когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера — Вилли обусловлено микроделецией, остальные — дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Патогенез
Патогенез синдрома Прадера — Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков.
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера — Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может играть роковую роль в развитии злокачественных новообразований у лиц с синдромом Прадера — Вилли.
Симптомы
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание.
В течение заболевания можно выделить две фазы: первая — свойственна детям 12-18 мес жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов — Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая — наступает позже, через несколько недель или месяцев. Появляются полифагия, постоянное чувство голода, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей.
Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает. Стопы и кисти больных диспропорционально маленькие — акромикрия. У детей отмечается гипогонадизм (у мальчиков — гипоплазия полового члена, мошонки, крипторхизм, а у девочек — недоразвитие половых губ и в 50% случаев — матки).
Рост больных нередко снижен. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед. ). Речь затруднена, словарный запас уменьшен. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, страбизм.
Встречаются и другие аномалии: микродонтия, гипоплазия хрящей ушных раковин, сколиоз, эктропион (выворот века), глаукома.
Нередко развитие сахарного диабета, который с возрастом имеет тенденцию к улучшению.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Лечение
Терапия синдрома Прадера — Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
Источник
Синдром Пра́дера — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин[2].
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat, когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13[3].
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер (A. Prader), Х. Вилли (H. Willi) и А. Лабхарт (A. Labhart)[4].
Особенности[править | править код]
Для синдрома Прадера — Вилли характерны:
- до рождения — низкая подвижность плода, часто — неправильное положение плода;
- дисплазия тазобедренных суставов;
- ожирение; склонность к перееданию (чаще проявляется к двум годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм) и в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.
Диагностика[править | править код]
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом).
Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Лечение[править | править код]
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Показан приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов).
Гипогонадизм обычно проявляется в микропении (микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков (крипторхизм); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск[править | править код]
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
Перспективы развития[править | править код]
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития.
Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание.
Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина. Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса[6]
См. также[править | править код]
- Синдром Мёбиуса
- Синдром Ангельмана
Примечания[править | править код]
Ссылки[править | править код]
- Л. З. Казанцева, П. В. Новиков, А. Н. Семячкина, Е. А. Николаева, М. Б. Курбатов, Э. В. Добрынина. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении. Московский НИИ педиатрии и детской хирургии Минздрава РФ / Рос. вестник перинатологии и педиатрии. — 1999. — № 4. — С.40-44.
- Сайт «Биология человека»
- Однородительская дисомия на сайте «Биология человека»
- О синдроме на сайте «Эндокринология» (EURODOCTOR.ru — Элитное лечение в Европе)[неавторитетный источник?]
- Форум родителей детей с этим диагнозом
- Мэтт Ридли. Геном: автобиография вида: В 23 главах. Глава из книги (Matt Ridley. Genome: The Autobiography of a Species in 23 Chapters)
- Подборка статей по теме, общение родителей (форум)
- Польза и вред от гормонов роста (реферат)[неавторитетный источник?]
- Кармен Сапиенца. Геномный импринтинг // Scientific American. Издание на русском языке. — 1990. — № 12. — С.14-20.
Источник

Синдром Прадера-Вилли – это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 – 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Синдром Прадера-Вилли
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% – наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом – полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек – недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения – ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза – показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации – основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития – синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция – увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии – бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия – прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина – карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике – предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
Источник