Код по мкб липодистрофия

Содержание
- Описание
- Причины
- Симптомы
- Лечение
Названия
Инсулиновая липодистрофия.
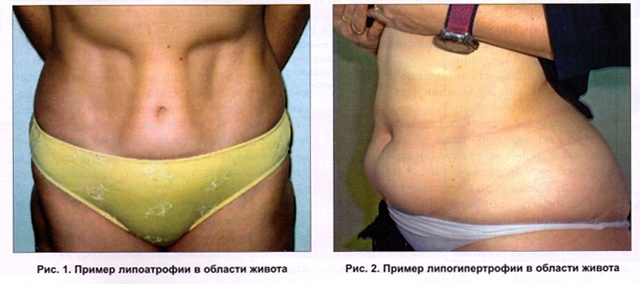
Инсулиновая липодистрофия
Описание
Инсулиновые липодистрофии — одно из осложнений инсулинотерапии, проявляющееся в виде атрофии (атрофическая форма) или гипертрофии (гипертрофическая форма) подкожной основы в местах введения инсулина. Чаще страдают дети и женщины.
Причины
В основе развития липодистрофий зачастую лежат нейродистрофические нарушения в зоне инъекции, обусловленные влиянием механических, физико-химических и термических раздражителей, связанных с нарушением процедуры инъекции и свойствами применяемого инсулина. Определенную роль в генезе липодистрофий отводят иммунологическим факторам, зависящим от качества инсулина. Это подтверждается значительным уменьшением случаев липодистрофий при использовании высокоочищенных инсулинов с нейтральной реакцией.
Липодистрофия развивается в различные сроки инсулинотерапии (от 1 мес до нескольких лет). Глубина поражения варьирует от небольших углублений до полного исчезновения подкожной основы на значительной поверхности. Проявление липодистрофии, кроме косметического дефекта, сопровождается нарушением всасывания инсулина, что может вести к декомпенсации сахарного диабета. Инъекции могут становиться болезненными, что особенно негативно у детей.
Симптомы
Липодистрофия проявляется, как правило, в виде атрофии подкожной основы в местах инъекций инсулина. Реже наблюдается гипертрофическая форма — образование своеобразной липомы, плотного жирового комочка в местах введения инсулина. Иногда липодистрофия образуется на местах, близких к участкам инъекций, — реперкуссионная липодистрофия.
Лечение
Профилактика и лечение при данном виде осложнений инсулинотерапии направлены на уменьшение факторов риска, способствующих возникновению липодистрофии. В целях максимального уменьшения механических, термических, химических раздражающих факторов, воздействующих на один и тот же участок кожи, нужно строго соблюдать правила введения инсулина. Необходимо использовать инсулин только комнатной температуры, ни в коем случае из холодильника. В настоящее время уже не используется протирание кожи спиртом перед инъекцией. Если же это происходит, необходимо подождать, пока спирт высохнет, и только затем делать инъекцию. Обязательна смена мест введения инсулина, что предполагает инъекцию в один и тот же участок тела не чаще 1 раза в 60 дней. Инъекции следует делать разовыми пластиковыми шприцами или шприц-ручками с острыми тонкими иглами, что делает процедуру атравматичной. При выполнении правил техники инъекций и использовании человеческого или свиного монокомпонентного инсулина с нейтральной реакцией рН, липодистрофии практически не развиваются.
Если липодистрофии уже возникли, необходимо проанализировать, какие правила введения инсулина нарушаются. Иногда просто смена места инъекции приводит к исчезновению липодистрофии. Рекомендуется введение человеческого или свиного инсулина в участки здоровой ткани, граничащие с липодистрофией. В места липодистрофий вводить инсулин нельзя, так как в этих участках нарушается его всасывание, что вызывает декомпенсацию заболевания. Иногда рекомендуется введение инсулина с раствором новокаина. В целях блокады источников раздражения и восстановления нервной трофики рекомендуют смешивать всю дозу инсулина или часть ее с равным по объему 0,5% раствором новокаина (или соответствующим эквивалентом 2% раствора новокаина). Такое обкалывание проводят длительно в зависимости от размеров участка липодистрофий. Новокаин можно назначать и в виде электрофореза на участки поражения. Для увеличения проницаемости тканей и ускорения всасывания инсулина применяют обкалывание участков липодистрофий лидазой или электрофорез с лидазой.
Обычно перечисленных мероприятий достаточно для профилактики и лечения липодистрофий.
Источник
Рубрика МКБ-10: E88.1
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E70-E90 Нарушения обмена веществ / E88 Другие нарушения обмена веществ
Определение и общие сведения[править]
Первичная липодистрофия
Первичные липодистрофии представляют собой гетерогенную группу очень редких заболеваний, характеризующуюся генерализованной или локальной потерей подкожно-жирового слоя в организме (липоатрофия).
Распространенность первичной липодистрофии оценивается менее чем 1/100 000.
Этиология и патогенез[править]
Генетические формы генерализованной липодистрофии (синдром Берардинелли-Сейпа) в большинстве случаев возникают в результате рецессивных мутаций в одном из двух генов — BSCL2, кодирующий сипин, либо BSCL1, кодирующий ацилтрансферазу (AGPAT2), участвующую в синтезе триглицеридов.
Этиология приобретенной генерализованной липодистрофии (синдром Лоуренса, синдром Сейпа- Лоуренса) неизвестна, но синдрому иногда сопутствуют признаки аутоиммунных заболеваний.
Парциальные липодистрофии могут быть семейными с доминирующей передачей. Гетерозиготные мутации были идентифицированы в гене LMNA, кодирующим ламин A/C ядер или в гене PPARG, кодирующем адипогенный транскрипционный фактор PPARgamma.
Некоторые менее типичные формы липодистрофии, сочетающиеся с признаками преждевременного старения, связаны с мутациями в генах LMNA или ZMPSTE24, кодирующем протеазу, ответственную за созревание преламина А в ламин А.
Приобретенная парциальная липодистрофия (синдром Барракера-Симонса) характеризуется потерей цефалоторакальной жировой ткани. Его этиология неизвестна, но мутации в гене LMNB2, кодирующие белок ламин B2, могут представлять собой факторы восприимчивости.
В настоящее время, высокоактивная антиретровирусная терапия (ВААРТ) для лечения ВИЧ-инфекции — является наиболее частой причиной приобретенных вторичных липодистрофических синдромов.
Клинические проявления[править]
В некоторых формах липоатрофия сочетается с селективной гипертрофией других жировых отложений. Часто присутствуют клинические признаки резистентности к инсулину: акантокератодермия (acanthosis nigricans), симптомы гиперандрогенизма. Все липодистрофии ассоциированы с дисметаболическими нарушениями с инсулинорезистентностью, изменением толерантности к глюкозе или диабетом и гипертриглицеридемией, что приводит к риску острого панкреатита. Диабет приводит к развитию хронических осложнений, затрагивающих сетчатку, почки, нервную и сердечно-сосудистую систему, а также стеатоза печени, что может привести к циррозу.
Липодистрофия, не классифицированная в других рубриках: Диагностика[править]
Генетическая диагностика проводится в специализированных лабораториях, в наиболее тяжелых формах может быть предложена антенатальная диагностика.
В подкожной жировой ткани отмечают тотальную атрофию, обнаруживают молодые (незрелые) липоциты. В органах системы кровообращения — склероз стенок сосудов, склероз стромы миокарда (в том числе с повреждением проводящей системы сердца), гипертрофия миокарда. Скелетные мышцы гипертрофированы. В печени — жировая дистрофия, острый и хронический гепатит. В ЦНС — атрофия и дистрофия нейронов мезо- и диэнцефальной области головного мозга. В яичниках развиваются склерокистоз, а также гипертрофия и гиперплазия воротных (лейдиговских) клеток яичников. Кроме того, отмечают системный фиброз стромы паренхиматозных органов, склероз сухожилий и капсул.
Дифференциальный диагноз[править]
Липодистрофия, не классифицированная в других рубриках: Лечение[править]
Лечение диабета, дислипидемии и возникающие осложнений — классические стратегии вмешательства. Полезны инсулин-сенсибилизирующие препараты. Терапевтические исследования рекомбинантного лептина человека у пациентов с очень низким уровнем лептина показали хорошие результаты в отношении метаболических и печеночных изменений.
Прогноз
Прогноз связан с ранним развитием и тяжестью диабетических, сердечно-сосудистых и печеночных осложнений.
Профилактика[править]
Прочее[править]
Очаговая липодистрофия
Синонимы: локализованная липодистрофия, парциальная липодистрофия
Определение и общие сведения
Очаговая липодистрофия характеризуется потерей подкожно-жирового слоя на небольших участках тела. Этот термин охватывает гетерогенную группу состояний. Липодистрофия часто бывает вызвана плохой техникой инъекции инсулина.
Этиология и патогенез
Очаговая липодистрофия также может быть ятрогенной этиологии, вследствие инъекций стероидов, соматостатина, глатирамера ацетата и др. Иногда патология вызвана давлением или является следствием панникулита, например шаровидного негнойного панникулита (синдром Вебера-Крисчена).
Болезнь Деркума (Adiposis dolorosa) является редкой патологией, которая характеризуется появлением болезненных жировых отложений, как правило, в период менопаузы у женщин с ожирением. Склеродермиформная липодистрофия живота и бедер может быть вызвана реакцией трансплантат против хозяина (РТПХ) после аллотрансплантции костного мозга. Идиопатические причины также нередки.
Дифференциальный диагноз
Дифференциальный диагноз включает в себя склеродермию, идиопатическую кожную атрофию, идиопатическую атрофодермию Пазини и Пьерини, генерализованную атрофию Гауэрса.
Лечение
Лечение должно способствовать устранению причины возникновения локализованной липодистрофии: модификация метода инъекции при инсулинотерапии, пластической хирургии, при необходимости — иммуносупрессивная терапия в случае реакции трансплантат против хозяина. Локализованная липодистрофия, связанная с инсулиновой терапией, как правило, проходит в течение нескольких недель после изменения места введения или изменения длины иглы.
Прогноз
Прогноз зависит от причины, но является более серьезным в случае панникулита или реакции трансплантат против хозяина.
Источники (ссылки)[править]
«Патологическая анатомия [Электронный ресурс] : национальное руководство / Под ред. М.А. Пальцева, Л.В. Кактурского, О.В. Зайратьянца — М. : ГЭОТАР-Медиа, 2011. — (Серия «Национальные руководства»).» — https://www.rosmedlib.ru/book/ISBN9785970419922.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Метилпреднизолон
- Метрелептин
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Названия
Название: Врожденная генерализованная липодистрофия.

Врожденная генерализованная липодистрофия
Описание
Врожденная генерализованная липодистрофия. Группа гетерогенных наследственных заболеваний, которые объединяет нарушение метаболизма в жировой ткани вплоть до ее полной атрофии. Симптомами этого состояния являются исчезновение подкожной жировой прослойки на большей части тела, черный акантоз кожных складок, сахарный диабет. Некоторые формы также сопровождаются разной степенью олигофрении, нарушением работы сердца, печени и скелетной мускулатуры. Диагностика врожденной генерализованной липодистрофии осуществляется на основании данных осмотра, семейного анамнеза, лабораторных анализов, а также методов генетической диагностики. Специфическое лечение отсутствует, применяют диетотерапию и симптоматическую терапию.
Дополнительные факты
Врожденная генерализованная липодистрофия (синдром Берардинелли-Сейпа) – несколько генетических состояний, при которых происходит атрофия жировой ткани с развитием некоторых сопутствующих симптомов. Практически одновременно было описано бразильским врачом-эндокринологом У. Берардинелли и норвежским педиатром М. Сейпом. Иногда можно встретить другое название врожденной генерализованной липодистрофии – синдром Сейпа-Лоуренса. Данное заболевание является относительно редким, встречаемость составляет порядка 1:10 000 000. Врожденная генерализованная липодистрофия с одинаковой частотой поражает как мальчиков, так и девочек и выявляется на первых месяцах жизни ребенка. Данные по летальности сильно разняться, по всей видимости, это обусловлено вариабельной выраженностью сопутствующих нарушений. Посредством методов современной генетики было определено несколько основных типов врожденной генерализованной липодистрофии, различающихся по генам, в которых произошли мутации.
Врожденная генерализованная липодистрофия
Причины
В общем плане причины развития врожденной генерализованной липодистрофии сводятся к нарушению метаболизма в жировой ткани (в основном, подкожной клетчатке), обусловленному генетическими мутациями. В результате этого происходит атрофия крупнейшего жирового депо в организме, что, в свою очередь, расстраивает весь липидный обмен в целом; а поскольку метаболизм представляет собой сбалансированную систему, то нарушения затрагивают также обмен углеводов и белков. При врожденной генерализованной липодистрофии у больных наблюдается затрудненное усвоение липидов, поэтому повышается их уровень в крови и нагрузка на органы, участвующие в их метаболизме. В первую очередь, это печень, кроме того, происходит аномальное накопление жира в других органах – почках, миокарде, скелетных мышцах, что приводит к расстройству их функций.
Врожденная генерализованная липодистрофия типа 1 обусловлена мутацией гена AGPAT2, расположенного на 9-й хромосоме. Он кодирует особый белок-фермент 1-ацилглицерол-3-фосфат-О-ацилтрансферазу 2, отвечающий за важный этап метаболизма жиров (фосфорилирование и старт образования фосфолипидов). Снижение активности фермента в результате дефектной структуры гена AGPAT2 приводит к нарушению усвоения липидов подкожной жировой клетчаткой, кроме того, это делает мембраны адипоцитов нестабильными из-за дефицита фосфолипидов. В результате всего этого развивается атрофия жировой ткани, происходит повышение уровня триглицеридов в крови, возникает отложение липидов в других органах, нарушается углеводный обмен с развитием сахарного диабета 2-го типа. Это и составляет клиническую картину врожденной генерализованной липодистрофии.
Форма заболевания 2-го типа развивается в результате мутации гена BSCL2, который расположен на 11-й хромосоме. Он кодирует белок под названием сейпин, поэтому механизм развития врожденной генерализованной липодистрофии в этом случае совсем иной. Сейпин контролирует развитие и дифференцировку адипоцитов, поэтому дефекты его структуры, обусловленные мутацией BSCL2, приводят к нарушению этих процессов. Из-за этого формирование подкожной жировой клетчатки затрудняется на самых ранних этапах развития. Врожденная генерализованная липодистрофия типа 3 обусловлена мутацией гена CAV1, локализованного на 7-й хромосоме. Продуктом его экспрессии является протеин кавеолин-1, принимающий активное участие в формировании кавеол – мембранных структур в виде инвагинаций и пузырьков, которые содержат на себе множество рецепторов и ферментов. Кавеолы принимают участие в поглощении липидов, формировании внутриклеточных липидных капель и межклеточных связей между адипоцитами. Нарушение структуры кавеолина-1 приводит к затруднению всех этих процессов и развитию врожденной генерализованной липодистрофии.
Врожденная генерализованная липодистрофия типа 4 вызывается мутацией гена PTRF, расположенного на 17-й хромосоме. Продукт данного гена, одноименный белок (polymerase and transcript release factor), является одним из факторов терминации трансляции, контролирующих образование ряда других протеинов, в частности, компонентов кавеол (белки кавеолин 1 и 3), поэтому механизм нарушений при этой форме патологии сходен с заболеванием 3-го типа. Особенностью мутации гена PTRF является тот факт, что страдают не только адипоциты, но также клетки скелетной мускулатуры и миокарда, поэтому эта форма патологии чаще других сопровождается мышечными нарушениями.
Классификация
Все формы врожденной генерализованной липодистрофии являются аутосомно-рецессивными наследственными заболеваниями, различаются лишь генетические и молекулярные механизмы развития нарушений. Небольшие различия имеются и в симптоматической картине патологии, однако они слишком стертые и непостоянные, чтобы по ним надежно разделить клинические формы синдрома Берардинелли-Сейпа. Поэтому генетическая классификация на сегодняшний день остается единственно верной и точной, согласно ей выделяют 4 основные типа этого заболевания:
Врожденная генерализованная липодистрофия тип 1. Обусловлена мутацией гена AGPAT2, расположенного на 9 — й хромосоме. Это приводит к дефекту структуры фермента 1-ацилглицерол-3-фосфат-О-ацилтрансферазы 2 адипоцитов, что и является причиной развития заболевания.
Врожденная генерализованная липодистрофия типа 2. Вызывается мутацией гена BSCL2, локализованного на 11 — й хромосоме. В результате этого возникают дефекты в структуре белка сейпина, отвечающего за процессы дифференцировки адипоцитов и развитие жировой клетчатки.
Врожденная генерализованная липодистрофия типа 3. Причина этой патологии заключается в мутации гена CAV1. Он находится на 7-й хромосоме и кодирует белок кавеолин-1, отвечающий за формирование на поверхности адипоцитов особых образований – кавеол. Нарушение их структуры приводит к расстройству метаболической активности жировой ткани, причем не только в подкожной клетчатке, но и в костном мозге, что может иметь последствия для системы гемопоэза.
Врожденная генерализованная липодистрофия типа 4. Вызвана мутацией гена PTRF, продуктом которого является один из белков — регуляторов трансляции. Этот ген расположен на 17-й хромосоме и посредством белка контролирует синтез нескольких других протеинов – в том числе и кавеолинов 1 и 3.
Симптомы
Наиболее характерным и явным симптомом врожденной генерализованной липодистрофии является почти полная атрофия подкожной жировой клетчатки, в результате чего достаточно четко контурируются подлежащие структуры. Определить заболевание может врач-педиатр сразу после рождения ребенка или в первые месяцы его жизни. В дальнейшем, помимо истончения подкожного жирового слоя, к симптомам присоединяются повышенный аппетит, быстрый рост, более крупные относительные размеры стоп, кистей, нижней челюсти, в ряде случаев наблюдаются также курчавые темные волосы. После пубертатного периода нередко отмечается более крупный размер половых органов (полового члена у мальчиков и клитора у девочек). Несмотря на отсутствие жировой клетчатки, больные врожденной генерализованной липодистрофией редко выглядят истощенными из-за выраженного развития мускулатуры (за исключением заболевания 4-го типа).
Умственная отсталость незначительной степени является непостоянным симптомом врожденной генерализованной липодистрофии, чаще всего она наблюдается при 2-м типе заболевания. На коже в области шеи и подмышечных впадин обнаруживается черный акантоз. Симптомы со стороны печени – боли в правом подреберье, изредка желтуха – являются признаком прогрессирования патологии и требуют симптоматической терапии. Кроме того, возможны проявления поражения почек – повышенный диурез, боли в пояснице, вплоть до развития хронической почечной недостаточности. У женщин возможно развитие вирильного синдрома с нарушениями менструаций, гирсутизмом, появлением акне и бесплодием.
Диагностика
Диагностирование врожденной генерализованной липодистрофии чаще всего производится педиатром, так как симптомы заболевания можно заметить при рождении или в первые месяцы жизни ребенка. Основными методами диагностики являются физикальное обследование, лабораторные тесты крови и мочи, изучение наследственного анамнеза пациента и генетические анализы. При осмотре выявляется практически полное отсутствие подкожной жировой клетчатки на всей поверхности тела, выраженное контурирование мышц, которые в большинстве случаев достаточно развиты. Характерный внешний вид больного – худое «скелетированное» лицо на фоне развитой фигуры, заметные подкожные вены, курчавые темные волосы, увеличенный размер стоп и кистей рук (акромегалия), участки черного акантоза в области кожных складок.
Пальпация при врожденной генерализованной липодистрофии может выявить увеличение печени и иногда селезенки, что подтверждается ультразвуковыми исследованиями. В тяжелых случаях возникает цирроз печени с его характерными проявлениями – желтухой, асцитом, отеками. Биохимический анализ крови выявляет гипертриглицеридемию, гипергликемию, повышенный уровень инсулина, глюкозотолерантный тест показывает резкое снижение всасывания углеводов тканями. Также в крови определяется общее повышение уровня липидов (гиперлипидемия), в основном за счет фракции липопротеидов очень низкой плотности (ЛПОНП). Если уровень глюкозы превышает 10 ммоль/л, то к проявлениям врожденной генерализованной липодистрофии присоединяются глюкозурия и повышение диуреза. Исследование почек, в том числе и ультразвуковое, может выявить нарушение их функций вплоть до хронической почечной недостаточности.
Исследование сердечно-сосудистой системы при врожденной генерализованной липодистрофии включает в себя электрокардиографию, эхокардиографию, определение артериального давления. В ряде случаев наблюдается увеличение размеров сердца (кардиомегалия), которое может с годами смениться дилатационной кардиомиопатией с развитием сердечной недостаточности. Увеличение равномерно затрагивает все отделы миокарда, что легко увидеть при ультразвуковом исследовании сердца, на ЭКГ будет определяться расширение комплекса QRS и нарушение реполяризации миокарда. Врожденная генерализованная липодистрофия также может проявляться аритмиями различного типа, возможна внезапная сердечная смерть. Артериальное давление обычно повышено, это может быть обусловлено поражением миокарда или же патологией почек.
При изучении наследственного анамнеза пациента можно выявить случаи врожденной генерализованной липодистрофии среди его родственников и определить аутосомно-рецессивный характер наследования. Генетическая диагностика, проводимая врачом-генетиком, основывается на прямом секвенировании последовательностей ассоциированных с заболеванием генов (AGPAT2, BSCL2, CAV1 и PTRF) для выявления мутаций.
Дифференциальная диагностика
Дифференциальную диагностику врожденной генерализованной липодистрофии следует проводить с приобретенными разновидностями этого состояния, анорексией, болезнью Иценко-Кушинга и тиреотоксикозом.
Лечение
Специфического лечения этого состояния не существует, возможна лишь симптоматическая терапия для уменьшения проявлений заболевания и предупреждения развития осложнений. С целью снижения нагрузки на организм при врожденной генерализованной липодистрофии разрабатывается специальная диета с пониженным содержанием жиров, которой больной должен придерживаться на протяжении всей жизни. Традиционные гиполипидемические средства (например, аторвастатин) при этом заболевании в ряде случаев являются неэффективными. Также в терапии этого состояния используют гепатопротекторы, гипогликемические средства (меглитиниды, бигуаниды), средства против аритмии – при наличии показаний. При умственной отсталости необходима работа с психологом и занятия с ребенком по особой программе. Некоторые больные врожденной генерализованной липодистрофией во взрослом возрасте решаются на разнообразные косметологические процедуры и пластические операции для коррекции чрезмерно худого лица.
Прогноз
Прогноз заболевания довольно часто неопределенный и во многом зависит от вовлеченности сердца, печени и почек в патологический процесс. В тяжелых случаях может развиваться сердечная недостаточность (в том числе и внезапная сердечная смерть), цирроз печени, ХПН. Возможны и диабетические нарушения – снижение микроциркуляции в конечностях, сетчатке глаза, более длительное заживление ран. У женщин в ряде случаев происходит поражение яичников (например, поликистоз), ведущее к бесплодию. При незначительно выраженных нарушениях вышеуказанных органов прогноз для жизни чаще всего благоприятный.
Источник