Код мкб синдром ретта

Рубрика МКБ-10: F84.2
МКБ-10 / F00-F99 КЛАСС V Психические расстройства и расстройства поведения / F80-F89 Расстройства психологического развития / F84 Общие расстройства психологического развития
Определение и общие сведения[править]
Синдром Ретта
Синдром Ретта представляет собой тяжелое расстройство нейроразвития, поражающее центральную нервную систему. Синдром Ретта наследуется как Х-сцепленный доминантный признак.
Синдром Ретта выделили из группы недифференцированного аутизма, или ранней злокачественной шизофрении (Rett A., 1963). Помимо Ретта, большой вклад в его изучение внесли японский исследователь A. Ishikava (1976, 1978) и особенно шведский учёный B. Hagberg (1993), которые осуществили клинико-психопатологическое описание синдрома Ретта, отметив стадийность течения, «моющие» движения кистей рук и другие моторные стреотипии. Синдром Ретта в течение последних двух десятилетий активно изучали во всём мире, о чём свидетельствует создание Международной ассоциации синдрома Ретта в 1980 г. Ряд авторов в рамках этого синдрома выявили тяжёлые неврологические расстройства, утрату способности к хватанию, удерживанию предметов в руках, судорожные расстройства, снижение сенсомоторного развития до уровня ребёнка 4-6 мес.
До 1990 г. считали, что синдром Ретта возникает только у девочек. В последние годы появились публикации, в которых представлены единичные описания лиц мужского пола с синдромом Ретта.
Эпидемиология
В 1995 г. M. Campbell и J. Shay указали распространённость синдрома Ретта у женщин в пределах 1:10 000 до 1:15 000, подчеркивая, что синдром практически не встречается у мужчин. Распространённость в популяции синдрома Ретта составляет 1 на 10 000 девочек. Н.Л. Горбачевская и В.Ю. Улас (1997), представляя обзор, указывают, что, по современным данным, распространённость этого синдрома варьирует от 0,72:10 000 до 3,5:10 000. Случаи возникновения синдрома Ретта у мальчиков единичны (при этом речь идёт скорее о предполагаемом синдроме).
Этиология и патогенез[править]
Несмотря на идентификацию мутаций в X-сцепленном гене метилированного CpG-связывающего белка 2 (MECP2) у большинства пациентов с синдромом Ретта, этиология остается неясной. Совсем недавно мутации в двух других генах — циклинзависимой киназы 5 (CDKL5) и Netrin G1, были идентифицированы у пациентов с клиническим фенотипом, который сильно перекрывается с синдромом Ретта.
Патогенез
Опираясь на нейроморфологические данные, полученные при исследовании синдрома Ретта, выдвинута идея прерванного развития ЦНС с остановкой развития головного мозга. Она основана на том, что у больных отмечают замедление и остановку в росте и развитии головного мозга наряду с замедлением развития других систем.
Предположение об атрофии паренхимы головного мозга отчасти подтверждено данными МРТ. При синдроме Ретта установлено снижение числа шипиков на дендритах и нарушение структуры дендритного дерева нейронов коры и базальных ганглиев головного мозга, которые, скорее, могут свидетельствовать о патологии межклеточных (синаптических) связей, что, по всей вероятности, поддерживает гипотезу о преимущественном нарушении развития головного мозга, а не его атрофии. Этой гипотезе также соответствуют данные о снижении плотности глутаматных рецепторов в базальных ганглиях, дофаминергических нейронов в хвостатом ядре, снижении функции холинергической системы. В связи с гипотезой о «прерванном развитии» особый интерес при синдроме Ретта представляют результаты исследования фактора роста нервов. Выявлено снижение его уровня в цереброспинальной жидкости и повышение титра аутоантител к этому фактору в крови обследованных. По этой причине появились предположения о существенной роли в генезе синдрома Ретта нейротрофических факторов.
Таким образом, патогенетические механизмы синдрома Ретта до конца не ясны.
Клинические проявления[править]
Синдром Ретта обычно возникает у детей с нормально протекавшими беременностью и родами. Он развивается в течение первых месяцев жизни. Пик болезни в среднем приходится на возраст 16-18 мес. В течение синдрома Ретта выделяют четыре стадии.
Стадии течения синдрома Ретта
1. I стадия («аутистическая»). В этом периоде происходит остановка психического развития ребёнка и накопления новых навыков, возникают снижение или исчезновение интересов к играм и окружающим близким родственникам. Нарастает чувствительность с чертами раздражительности, меняется настроение, становясь неустойчивым и сниженным, с чертами капризности.
В последующем становятся отчётливыми расстройства настроения астенодистимического типа, перемежающиеся при их усилении ажитированными состояниями. Больные утрачивали интерес к игрушкам, не могли организовать даже примитивную игру, проводили время в бездействии, бесцельно перемещались по помещению.
Длительность этой стадии может составлять от нескольких месяцев до полутора лет. В целом поведение ребёнка на этом этапе почти не отличается от того, что наблюдают при других аутистических синдромах. Из особых признаков обращают на себя внимание частичная сохранность глазной реакции, тактильного контакта с матерью, понижение мышечного тонуса и замедление роста головы.
2. II стадия («быстрый регресс»). Аутистические расстройства смягчаются, быстро наступает распад речи, возникают апраксия и общее моторное беспокойство. В кистях рук появляются движения «моющего» и «потирающего» характера. Больные царапают себя, жуют пальцы рук. Могут возникать и другие насильственные движения: касания руками подбородка, головы, груди; нанесение сведёнными в кулаки пальцами рук ударов по щекам, подбородку, заведение рук за спину и др. Перечисленные движения больные совершают непрерывно, реже — через небольшие интервалы насильственно (за исключением периода сна или при фиксации рук).
Постепенно пропадает способность к удерживанию в руках предметов (например, вложенные в руки игрушки, ложка тут же выпадают). В связи с постоянными движениями «потирающего» характера в области II фаланги первого пальца, а также II и III фаланг второго пальца развиваются гипертрофия мышц, мозолистые утолщения и экскориация кожных покровов; в области IV и V пальцев рук, пястных костей — истончение и атрофия мышц. У детей меняется походка: они широко расставляют ноги при ходьбе и двигаются покачиваясь (эти изменения напоминают атаксическую походку).
Отмечают мышечную дистонию, временами переходящую в атонию или гипертонию; лёгкую атрофию мышц стоп и голеней (по типу «носков»), синюшность кожных покровов и холодность их на ощупь, а также рекурвацию в суставах, приводящую к частому спотыканию и трудностям спуска с лестницы. Движения туловища обеднены, голова втягивается в плечи. Гипертонус шейных и плечевых мышц сочетается со сколиозом и кифосколиозом. При прогрессировании описанных нарушений дети полностью теряют способность к изменению положения тела (из положения лёжа не могут сесть или встать).
На этой стадии больные выглядят отгороженными от внешнего мира. Сохраняющаяся реакция слежения глазами крайне истощаема; появляются гримасничание, нахмуривание бровей, зажмуривание глаз. Выраженность аути-стического отрешения изменчива, и временами появляется лёгкая тенденция к общению, тогда же отмечают и определённую сохранность эмоционального отношения к родным. Но большую часть времени поведение остаётся однообразным и монотонным.
У больных на этой стадии могут возникать нарушения дыхания: учащённое дыхание сопровождается внезапным апноэ с напряжением мышц шеи и плечевого пояса. Иногда это состояние переходит в общее напряжение с приведением рук к телу и нередко сопровождается гортанным выкриком. Продолжительность этих состояний может составлять несколько секунд, после чего восстанавливается нормальное дыхание. Подобные состояния имеют тенденцию к повторению через разные промежутки времени. В некоторых случаях при подобном нарушении дыхания происходит заглатывание воздуха с развитием пневматоза кишечника.
Длительность стадии «быстрого распада» может составлять от нескольких недель до нескольких месяцев, реже лет.
3. III стадия («псевдостационарная»). Характерно слабоумие с полной утратой речи (сохраняются лишь контуры отдельных слов и слогов). Более длительное время сохраняется лишь понимание простейших жестов бытового содержания. Движения в кистях рук в этот период протекают с меньшим мышечным напряжением и возникают реже; появляется крупноразмашистый тремор головы, туловища, рук, усиливающийся при попытке выполнения направленных движений. Утрачивается даже способность жевать, возникает попёрхивание, затем нарушается глотание и больные переходят на сосание.
В 1/3 случаев у больных возникают эпилептиформные припадки (малые и большие судорожные). Могут также возникать атипичные приступы по типу вздрагиваний с ознобоподобными явлениями, дрожью всего тела во время сна. В ряде случаев наблюдают гиперкинез в группах мышц верхнего плечевого пояса в виде частых повторов, что напоминает приступы фокальной эпилепсии.
Общая активность детей быстро обедняется, приобретая характер бесцельной ходьбы взад и вперёд, во время которой может возникать мимолётный интерес к игрушкам. Больные подходят к ним, приближают к ним лицо, касаются губами.
Сохранены глазная реакция «глаза в глаза» и реакция откликания на зов, но они резко отставлены во времени и кратки. В отношениях с матерью и близкими ребёнку лицами сохраняется только «тактильная игра», свойственная детям первого года жизни с возможностью лёгкого эмоционального оживления.
Эта стадия может длиться несколько лет (иногда 10 лет и более), в течение которых отрешённость от внешнего мира и аутистические проявления нивелируются и даже появляется некоторое оживление интереса к окружающему. Одновременно несколько смягчается насильственность «моющих» и других движений в кистях рук.
4. IV стадия («тотальное слабоумие»). Возникает полная утрата не только речи, но и способности к ходьбе и жеванию, а также распад других навыков. Нарастают неврологические симптомы (развиваются спинальная атрофия, спастическая ригидность с наибольшей выраженностью в конечностях), которые на этой стадии преобладают в клинической картине. Смерть наступает в разные сроки (обычно через 12-25 лет от начала заболевания, что практически соответствует и возрасту больных).
Синдром Ретта: Диагностика[править]
Диагноз клинический, основан на диагностических критериях Треватана, недавно пересмотренных на встрече экспертной группы Европейского общества детской неврологии.
Для поставки диагноза типичного синдрома Ретта требуется наличие: периода регрессии с последующим восстановлением или стабилизацией, наличие всех основные критериев и наличие всех критериев исключения, наличие дополнительные критерии не требуются, хотя они часто присутствуют при типичном синдроме Ретта.
Основные критерии: частичная или полная потеря приобретенных целенаправленных движений рук,
частичная или полная потеря разговорного языка, нарушения походки, стереотипные движения рук.
Критерии исключения: повреждение головного мозга вследствие травмы, нейрометаболического заболевания или тяжелой инфекции, выраженное отклонение психомоторного развития в первые 6 месяцев жизни.
Дифференциальный диагноз[править]
Дифференциальный диагноз включает аутизм и синдром Ангельмана; катаракту, ретинопатию или атрофию зрительного нерва; перинатальное или постнатальное повреждения головного мозга, нарушения метаболизма или нейродегенеративные расстройства; приобретенное неврологическое расстройство из-вследствие тяжелой травмы головы или инфекции. Болезни накопления обычно исключается наличием органомегалии.
Синдром Ретта: Лечение[править]
Лечение в основном симптоматическое, сфокусировано на оптимизации способностей каждого пациента. Наиболее эффективен междисциплинарный подход (включая диетологов, физиотерапевтов, профессиональных терапевтов, логопедов и музыкальных терапевтов). Следует обратить внимание на сколиоз и развитие спастичности, а также на развитие эффективных коммуникационных стратегий. Психосоциальная поддержка семей имеет важное значение. Фармакологические подходы направлены на коррекцию нарушений сна, нарушений дыхания, судорог, стереотипных движений и общего благополучия.
Профилактика[править]
Поскольку пациенты с синдромом Ретта имеют повышенный риск угрожающих аритмий, связанных с удлинением интервала QT, рекомендуется избегать приема ряда препаратов. Клиническая картина развивается поэтапно в течение ряда лет, а прогноз оставляет желать лучшего.
Прочее[править]
Атипичный синдром Ретта
Определение и общие сведения
Атипичный синдром Ретта представляет собой психомоторного расстройство, которое диагностируется, когда ребенок обнаруживает симтоматику типичного синдрома Ретта, но не выполняет все его диагностические критерии. По данным литературы, до 32% случаев синдрома Ретта демонстрирует нетипичный фенотип.
Распространенность атипичного синдрома Ретта оценивается на уровне около 1/45000. Как и классический синдром Ретта, атипичная вариант преимущественно поражает девочек.
Клинические проявления и этиология
Были выделены несколько субвариантов атипичного синдрома Ретта:
1) Тип с ранним началом судорог (вариант Hanefeld) характеризуется наличием судорожных припадков в течение первых месяцев жизни с последующим развитием симтоматики типичного синдрома Ретта. Данный вариант часто вызывается Х-сцепленной мутацией гена CDKL5 (Xp22). Транслокации с участием гена NTNG1 (1p13.2-P13.1) также была идентифицированы у пациента с ранними приступами и атипичным фенотипом.
2) Врожденный вариант (вариант Роландо) является наиболее тяжелой формой атипичного синдрома Ретта, с клиникой классического варианта в течение первых трех месяцев жизни. Этот вариант, как правило, вызывается мутациями гена FOXG1 (14q11-q13). Синдром микроделеции 14q12 также была описан в рамках этого варианта, но дополнительно присутствовал дисморфизм лица.
3) Вариант “forme fruste” (абортивная форма) представляет собой более мягкий вариант с началом в раннем детстве и неполным и затяжным течением.
4) Вариант с поздней детской регрессией характеризуется нормальной окружностью головы и более постепенным и поздним началом (конец детства) с регрессом речевых и двигательных навыков.
5) Вариант сохранной речи (вариант Zappella) отмечен восстановлением некоторых словесных и мануальных навыков и вызывается по крайней мере в некоторых случаях мутации гена MECP2 (Xq28), который также отвечает за большинство случаев классического синдрома Ретта.
Диагностика
Диагноз основывается на клинической оценке, с использованием диагностических критериев, первоначально сформулированных Hagberg в 1994 году: нетипичный случай должен отвечать по крайней мере трем из шести основных критериев и ,по меньшей мере, пяти из одиннадцати вспомогательных. Молекулярный анализ может позволить подтверждить диагноз.
Дифференциальный диагноз
Дифференциальный диагноз включает синдром Ангельмана, аутизм, церебральный паралич, врожденные нарушения метаболизма и серьезного интеллектуального дефицита
Лечение
Лечение симптоматическое и поддерживающее. Лекарства могут быть использованы для коррекции нарушений дыхания, нарушений сна (мелатонин), возбуждения (рисперидон), агрессии (карбидопа, леводопа). Противоэпилептические препараты могут быть использованы для контроля судорог. Анти-рефлюкс средства также могут потребоваться.
Прогноз
Течение атипичного синдрома Ретта варьируется, в том числе возраст начала и тяжесть симптомов. Большинство людей доживают до среднего возраста и старше.
Источники (ссылки)[править]
Источники (ссылки): Источники (ссылки): Психиатрия [Электронный ресурс] / Под ред. Т.Б. Дмитриевой, В.Н. Краснова, Н.Г. Незнанова, В.Я. Семке, А.С. Тиганова — М. : ГЭОТАР-Медиа, 2011. — https://www.rosmedlib.ru/book/ISBN9785970420300.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Ретта.
Синдром Ретта
Описание
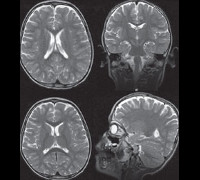
Синдром Ретта. Генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Дополнительные факты
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Причины
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Судороги. Эхолалия.
Диагностика
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник