Код мкб синдром крузона

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Крузона.
Синдром Крузона
Описание
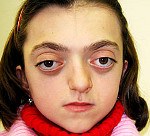
Синдром Крузона. Редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Дополнительные факты
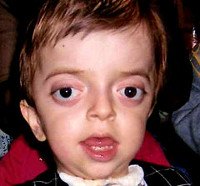
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Синдром Крузона
Причины
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Рвота. Судороги.
Диагностика
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник

Синдром Крузона – деформация лицевой и мозговой части черепа, формирующаяся в период внутриутробного развития и передающаяся по наследству. Патология возникает достаточно редко, протекает тяжело и сопровождается аномальным преждевременным заращением швов между черепными и лицевыми костями. Раннее закрытие черепных швов способствует ограниченному объему черепа и изменению формы головы. У больных нос приобретает крючковидную форму, нарушается слух и зрение. При поражении коронарного шва, идущего от одного уха к другому, возникают аномалии глазниц – они становятся выпуклыми. У больных изменяется высота лба и выраженность челюстной мускулатуры. Косметические уродства часто сочетаются с задержкой интеллекта или умственной отсталостью.
Краниофасциальный дизостоз характеризуется нарушением процессов окостенения черепа. Это заболевание впервые описал педиатр из Франции Крузон в 1912 году. С тех пор оно носит имя своего первооткрывателя. Патологическое состояние одинаково часто встречается как среди мальчиков, так и среди девочек. Точная частота возникновения данного недуга в настоящее время остается неизвестной.

первый пациент с описанным синдромом Крузона (1912 г.)
Синдром Крузона наследуется по аутосомно-доминантному механизму. В некоторых случаях причиной патологии может стать спонтанная мутация генов. У новорожденных детей имеются швы между костями лица и черепа. По мере развития детского организма череп расширяется благодаря этим швам. У здоровых людей они полностью зарастают лишь после остановки роста мозга и его полноценного формирования. При синдроме Крузона черепные кости срастаются намного раньше. Череп больных растет в сторону открытых швов, что приводит к появлению выраженных дефектов внешности.
Нередко деформация черепа сочетается с аномалиями отдельных структур опорно-двигательного аппарата — межпозвоночных сочленений, крупных суставов конечностей: локтевого, плечевого, коленного. Краниосиностоз осложняется развитием гидроцефалии — водянки головного мозга, приводящей к повышению внутричерепного давления. Тяжелая деформация лицевых костей сопровождается нарушением носового дыхания. Особенно опасно подобное расстройство для новорожденных детей, которые еще не умеют дышать ртом. Возможно появление ночного апноэ – внезапной остановки дыхания во сне.
Диагностика синдрома заключается в тщательном визуальном осмотре больного, проведении рентгенологического и молекулярно-генетического исследований. Синдром Крузона — неизлечимое заболевание. Для поддержания жизни больных на оптимальном уровне проводят симптоматическую терапию консервативного и хирургического характера. Краниопластика и другие корригирующие оперативные вмешательства устраняют деформации черепа.
Причины

Синдром Крузона — наследственная патология, возникающая вследствие мутационного процесса. Передается недуг по аутосомно-доминантному принципу от родителей детям. Когда в одной родительской хромосоме присутствует мутировавший ген, дети не всегда наследуют заболевание. В таких семьях риск рождения ребенка с синдромом Крузона составляет 50%. Причем новорожденные дети могут даже не быть носителями дефекта. Тщательное обследование на этапе планирования беременности позволяет повысить шанс рождения здорового малыша.
Данный недуг развивается при мутации гена, расположенной в X хромосоме. Этот ген является нестабильным из-за значительных размеров и большого количества экзонов. Его мутации приводят к развитию наследственных патологий скелета человека. Происходит нарушение структуры соединительной ткани, костей и хрящей. В области межкостных швов скапливаются фибробласты, синтезирующие межклеточное вещество, продуцирующие белки — коллаген и эластин и активизирующие процессы окостенения. Это основной этиопатогенетический фактор синдрома Крузона.
К факторам, способствующим развитию патологии, относятся: наличие патологии в семейном анамнезе, возраст отца старше 60 лет в период зачатия ребенка.
Известны случаи ненаследственного характера синдрома Крузона, развивающегося на фоне спонтанной мутации в гене. Чаще всего такие нарушения встречаются по отцовской линии.
Симптоматика
Клинические признаки синдрома Крузона весьма специфичны. Они становятся заметными сразу после рождения. Выраженность проявлений достигает своего максимума ближе к 3-4 годам.

- Основной симптом патологии – краниосиностоз, при котором прочно срастаются кости черепа по венечному или стреловидному шву. При этом рост головы останавливается, и ребенок приобретает характерные черты: дистальный прикус, большое расстояние между глазами, клювовидный нос, низкое расположение ушей, выступающий язык, укороченная и низко расположенная губа, недостаточное смыкание челюстей, редкие зубы, высокое небо, выдающийся вперед подбородок, расщелина на язычке. Характерная черта синдрома — неполное смыкание зубов.
- Дополнительные признаки – синдактилия пальцев, атрезия хоан, сколиоз, лордоз, мигрень, акантоз.
- По мере прогрессирования синдрома формируется короткоголовость – брахицефалия. Возможные виды деформации черепной коробки: клиновидная или ладьевидная голова, гидроцефалоидная деформация черепа в форме трилистника. При пальпации определяются уплощенные края швов.

- Поражение зрительного анализатора проявляется расходящимся косоглазием, нистагмом, эктопией, колобомой. Экзофтальм обусловлен незначительной глубиной орбит. У больных раскосые глаза постоянно вращаются, совершая спонтанные движения. Значительно снижается острота зрения. У 80% больных выявляют частичную атрофию зрительного нерва.
- Нарушения слуха вплоть до полной глухоты происходит при поражении пирамиды височной кости, фиксации и деформации слуховых косточек. У больных изменяется форма внутреннего слухового канала, понижается звуковая проводимость костей, развивается атрезия внешнего слухового хода.
- Поражение нервной системы – умственная отсталость, задержка речи, судороги. Нарушения интеллекта сохраняются на протяжении всей жизни, затрудняя межличностное общение в социуме.
Синдром Крузона — серьезный недуг, приводящий к развитию у ребенка различных осложнений.
Негативные последствия заболевания:
- гидроцефалия — водянка мозга,
- потеря зрения — результат сдавливания зрительного нерва и его атрофии,
- язвенный кератит развивается при неполном смыкании век и частичном высыхании роговицы,
- умственная неполноценность,
- трудности с адаптацией в обществе.
Диагностические мероприятия
Диагностика патологии включает следующие методики:
- общий осмотр позволяет обнаружить характерные признаки синдрома;
- сбор семейного анамнеза проводится с целью выявления случаев заболевания в роду;
- рентгенологическое исследование — оценка формы черепа и наличия сращений швов, расширение ямок гипофиза, уплощение глазницы со своеобразными вдавлениями;
- ядерно-резонансная или компьютерная томография — гиперостозы, аномалии слуховых косточек, отсутствие полости среднего уха, опущение миндалин мозжечка;
- генетическое обследование и молекулярная диагностика методом ПЦР — выявление мутации в 7 и 9 экзонах гена десятой хромосомы;
- осмотр у офтальмолога, определение остроты зрения, рефрактометрия;
- консультация ЛОР-врача и аудиометрия;
- работа с психотерапевтом или неврологом, позволяющая определить уровень психического развития больного.
Лечение
Синдром Крузона — неизлечимое заболевание, требующее функциональной и косметической коррекции. Достичь ее можно лишь оперативным путем.
Хирургическое вмешательство направлено на восстановление формы черепа и устранение синостозов. Такие операции проводят, начиная с рождения и до остановки роста черепной коробки. Они позволяют снизить уровень внутричерепного давления, исправить костно-мышечные деформации и предотвратить дальнейшее прогрессирование умственной отсталости, а также психоневрологических нарушений. Для этого раскрывают рано заращенные швы черепа и исправляют форму лица.

Основные виды операций:
- Краниопластика и вскрытие синостозированных швов – удаление и расширение фрагментов костей черепа для предотвращения повреждение мозга и исправления формы черепной коробки.
- Создание искусственного блефарофимоза для снижения выраженности экзофтальма и исправления положения глазного яблока.
- Расширение хоан для восстановления дыхания.
- Установка вентрикулоперитонеальной шунтирующей системы при гидроцефалии.
- Использование аппарата Илизарова для выдвижения вперед верхней челюсти и улучшения дыхательной функции.
- Коррекция выступающей нижней челюсти и нормализация ее внешнего вида.
- Радикальные комплексные операции направлены на устранение дефектов лица – исправление верхнечелюстной гипоплазии, восстановление зубного ряда.
В настоящее время внедряются в хирургическую практику дистракционные методики коррекции деформаций лицевых костей. С помощью специальных аппаратов для смещения любого участка черепа и исправления костных дефектов процесс лечения проходит быстрее и более эффективно.

постоперационные приспособления для улучшения эффективности лечения
В тех случаях, когда операция противопоказана ребенку, нарушение функции дыхания компенсируют постоянной кислородотерапией, дыханием под повышенным давлением, использованием воздуховодов, трахеостомией.
Облегчить состояние больного также поможет лекарственная терапия. Больным назначают:
- Ноотропы – «Пантогам», «Пирацетам», «Винпоцетин».
- Сосудистые препараты – «Кавинтон», «Циннаризин», «Актовегин».
- Диуретики – «Лазикс», «Верошпирон», «Диакарб».
Больные с синдромом Крузона находятся на учете у офтальмологов и оториноларингологов, которые при выявлении каких-либо отклонений назначают адекватную терапию. Дети с психическими расстройствами нуждаются в специальном лечении и занятиях с логопедом.
Прогноз
Прогноз при синдроме Крузона неоднозначный. Он зависит от выраженности черепной деформации и состояния мозговой ткани. Первоначальная реконструктивная операция проводится на первом году жизни. По мере роста ребенка она дополняется новыми вмешательствами на костях и мягких тканях лица.

Болезнь часто сопровождается задержкой психомоторного развития и судорожными приступами. Поэтому многие специалисты оценивают прогноз как неблагоприятный. Даже при проведении поддерживающей терапии в полном объеме у больных прогрессирует расходящееся косоглазие, развивается глухота и гипоплазия лица. Больные, вовремя обратившиеся за медицинской помощью и постоянно наблюдающиеся у специалистов, часто доживают до преклонных лет. Некоторые лица с синдромом Крузона все же сохраняют социальную адаптацию, несмотря на грубые косметические недостатки.
Причинами инвалидности являются нарушения зрения и слуха, а также умственная отсталость. Со временем дефекты костей становятся все более выраженными. Поскольку в настоящее время не разработаны профилактические мероприятия, предупреждающие развитие данного недуга, надеяться можно только на прогрессирование медицинских технологий, которые когда-нибудь смогут избавить людей от генетических расстройств.
Видео: мини-лекция о синдроме Крузона
Видео: фильм о синдроме Крузона
Источник