Код мкб синдром корнелии де ланге

Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
1. Какие системы органов поражаются
Для новорожденных характерно наличие респираторного дистрегс-синдрома из-за специфического строения носоглотки, поэтому в анамнезе у этих пациентов регистрируются частые инфекционно-воспалительные заболевания дыхательных путей
При вскрытии умерших больных обнаруживаются разнообразные дефекты головного мозга (недоразвитие нижней лобной извилины, расширение желудочков, дисплазия и гипоплазия извилин), гистология нередко показывает выраженную поперечную исчерченность нейронов внешнего зернистого слоя коры больших полушарий и расстройство топографии нейронов мозжечка.
Более чем в половине всех случаев амстердамскому нанизму сопутствуют дефекты в структуре сердца (аортолегочное окно; незаращенная перегородка, разделяющая как предсердия, так и желудочки, часто в комбинации с сосудистыми нарушениями; тетрада Фалло), дефекты в структуре ЖКТ (в основном, нарушения поворота кишечника), мочеполовой системы (кистозные образования почек, одиночные и множественные, иногда подковообразная почка и гидронефротические ее изменения, крипторхизм, двурогая матка)
Это заболевание, характеризующееся множеством дефектов развития, является по своей сути пока еще не раскрытой генетической аномалией, которая начинается в период формирования эмбриона. Процесс, запущенный патогенным фактором, продолжается и усугубляется в дальнейшем, после рождения ребенка.
2. Частота встречаемости в популяции
Заболевание встречается у новорожденных с частотой от 1:30000 до 1:10000 (от 1:10000 до 100000), соотношение мальчиков и девочек — 1:1. Тип наследования не уточнен, известны семейные случаи с аутосомно-рецессивным типом наследования. У большинства больных кариотип нормальный.
3. Тип наследования
Предполагается аутосомно-доминантное и X-сцепленное наследование. Большинство случаев спорадические. Почти в половине случаев причиной синдрома может быть мутация в гене NIPBL, картированном в 5p13., при Х-сцепленной форме выявлена мутация гена SNC1L1, локализованного на Xp11.22-p11.21. (т.е. синдром является генетически гетерогенным, обусловленным мутациями в гене №РВЬ, 8УС3, микродупликацией локусов q25-q29 хромосомы 3) Всего на данный момент известно более 400 случаев этого заболевания в разных странах.
4. Кем и когда впервые было открыто и описано данное заболевание
Первым сделал описание синдрома как самостоятельного заболевания немецкий врач В. Брахман в начале ХХ в. Несколько позже педиатр из Нидерландов Корнелия де Ланге (де Ланж) вела двух маленьких пациенток, страдающих этим заболеванием, и на материалах наблюдений подробно описала его. Эта патология может еще называться синдромом Брахмана де Ланге или дегенеративным нанизмом типа «Амстердам» (Амстердамская карликовость), т.к. трое детей с этим диагнозом жили в столице Нидерландов.
5. Этиология (причина возникновения):
В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов риска:
1. Наличие в семейном анамнезе этого синдрома, т.к. в этом случае (если предположение о рецессивном способе передачи гена верно) возможность появления следующего ребенка с патологией составляет 25%.
2. Степень вероятности повторения ситуации в одиночных эпизодах при отсутствии хромосомных мутаций у родителей теоретически равна 2 %.
3. Преобразования хромосом могут возникать вследствие тяжелых инфекций и интоксикаций, перенесенных будущей матерью в первые три месяца беременности, побочных действий химиотерапевтических лекарственных средств и некоторых физиотерапевтических процедур.
4. Генным мутациям могут способствовать эндокринные заболевания матери и радиация.
5. Солидный возраст отца ребенка, либо материнский возраст более 35 лет.
6. Мать и отец — кровные родственники.
6. Клиническая картина
В клинической картине синдрома выделяют следующие основные признаки:
• «причудливое лицо», а именно густой для новорожденного волосяной покров головы, соединенные брови и длинные загнутые ресницы, деформация ушей и маленький нос с открытыми спереди ноздрями, большой носогубный фильтр, тоненькая красная кайма верхней губы, опущенные уголки губ;
• микроцефалия головного мозга;
• брахицефалия (уменьшение высоты черепа с одновременным увеличением его горизонтального размера);
• патологии полости рта и носоглотки (атрезия хоан, аркообразное небо с расщелиной, сбои в процессе прорезывания молочных зубов);
• дисфункции зрения (страбизм, нарушения формы хрусталика, роговицы глаза, близорукость, атрофия зрительного нерва);
• укороченные конечности и другие их аномалии;
• «мраморный» рисунок кожи;
• задержка полового развития;
• гипертрихоз;
• эпизодическая судорожная готовность, гипотонус, гипертонус мышц;
• карликовость;
• умственная отсталость разной степени (от незначительных отклонений от нормы (редко) до олигофрении и имбецильности в большинстве случаев.
Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает на себя внимание маленький вес ребенка при рождении, который составляет 2/3 веса здорового ребенка, родившегося на аналогичном сроке беременности.
Описанные заболевания глаз – миопия, микрокорнеа, астигматизм, атрофия или колобома зрительного нерва, косоглазие, птоз.
*Вероятность развития дефектов в процентах:
Характерных и диагностически важных черепно-лицевых аномалий:
Микробрахицефалия (93%), низко расположенные ушные раковины (58%), синофриз (99%), тонкие чёткие брови, длинные загнутые ресницы (100%), запавшая переносица (95%), маленький нос с открытыми вперёд ноздрями (100%), атрезия хоан, длинный выступающий фильтр (94%), микрогения (97%), тонкая верхняя губа (94%), рот в виде полумесяца (94%), высокое арковидное нёбо (86%), позднее прорезывание зубов, большие промежутки между зубами (86%), иногда – расщелина нёба (30%).
Пороки конечностей: акромикрия (93%), ограничение подвижности локтевых суставов (64%), фокомелия и олигодактилия (30%), клинодактилия V пальца (69%),
*реже встречаются гипоплазия лучевой кости, короткие I метакарпальные кости.
Наблюдаются гипертрихоз (78%), мраморная кожа (60%), гипоплазия сосков (55%) [3]
7. Профилактика
Профилактикой синдрома, факторы возникновения которого точно не установлены заниматься сложно.
Однако, с учетом известных источников генных мутаций, можно рекомендовать в качестве профилактических мер:
предотвращение зачатия детей от матери и отца – кровных родственников;
тщательно обследоваться в случае возможности позднего материнства и отцовства;
беременным женщинам, избегать заражения вирусными инфекциями, особенно в первом триместре, а в случае заражения применять лекарственную терапию только по назначению врача.
Женщины и мужчины, в семейном анамнезе которых присутствует синдром Корнелии де Ланге, обязательно должны посетить медико-генетическую консультацию. Во время беременности женщинам обязательно нужно обследоваться на наличие протеина-А плазмы крови. [2]
8. Продолжительность жизни
При синдроме Корнелии де Ланге продолжительность жизни зависит от степени дефекта развития внутренних органов. Если аномалия несовместима с жизнью и своевременно не проведено оперативное вмешательство, смерть может наступить в течение месяца после рождения. Отсутствие соматических отклонений, ранняя диагностика дефектов, позволяющая их устранить, обеспечивает долгую жизнь пациентам. Прогноз ухудшается в случае вирусных инфекций, которым неспособна противостоять иммунная система. Обычный грипп может закончиться летальным исходом. В среднем дети с синдромом Корнелии де Ланге доживают до 13 лет. Фиксировались случаи при легкой, стертой форме, после адекватного устранения аномалий, когда пациент умирал в преклонном возрасте. [2] По некоторым источникам больные со стертой формой заболевания или удачно проведенными операциями по устранению дефектов развития иногда доживали до пятого-шестого десятка лет
9. Патогенез
В рамках фенотипической вариабельности выделяют два варианта синдрома: первый (классический) характеризуется выраженной пренатальной гипоплазией, значительной задержкой физического и умственного развития и грубыми пороками развития. Второй (доброкачественный) сопровождается аналогичными лицевыми и малыми скелетными аномалиями, пограничной задержкой психомоторного развития; крупные пороки развития отсутствуют .
10. Дифференциальный диагноз
Частичная трисомия 3q, синдром Фринса, фетальный алкогольный синдром.
11. Код по МКБ-10
Q87.1 Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью [2]
Список литературы:
[Электроный ресурс]: https://zdorovo.live/sustavy/sindrom-kornelii-de-lange-prichiny-formy-i-simptomy-metody-lecheniya-prognoz.html#i-17
[Электроный ресурс]: https://ilive.com.ua/health/sindrom-kornelii-de-lange_103875i88403.html
Козлова С.И. Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. — М.: КМК, Авторская академия; 2007.
Барашнев Ю.И., Бахарев В.П., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний и психоречевого развития. Врожденная гидроцефалия. Эпилептический синдром резидуального генеза.
Источник
Многие из школьных уроков биологии помнят, что наследственная информация, передающаяся от отца с матерью их детям, заключается в геноме человека, состоящем из 23 пар хромосом. В них содержатся около 28 тысяч генов, каждый из которых играет свою важнейшую роль в формировании человеческого организма. Мутационные изменения только одного из них могут спровоцировать синдром Корнелии де Ланге, очень неприятное, а в большинстве случаев и достаточно тяжелое заболевание, нередко приводящее к летальному исходу. Многие авторы утверждают, что у страдающих этим синдромом нет шансов. И тем не менее не стоит отчаиваться, потому что современные наука и медицина творят настоящие чудеса.
Этимология названия
Синдром Корнелии де Ланге был так назван в честь подробно его описавшего детского врача по имени Корнелия де Ланге, в 30-х годах ХХ века жившего и работавшего в Голландии. Она в своей практике наблюдала 5 подобных случаев, в последний раз сразу двух девочек, не являвшихся родственницами. В 1933 году Корнелия сделала подробное описание и заключение своих наблюдений. Но гораздо раньше (в 1916 году) это же заболевание диагностировал и подробно описывал немецкий врач В. Брахман, поэтому часто используют название синдром Брахмана — Ланге. Кроме того, встречается название амстердамский синдром, так как в этом городе зафиксировано сразу трое детей, у которых обнаружилась такая патология. Все три наименования – это одно и то же заболевание. Оно встречается на всех континентах, у людей всех рас и этнических групп, с одинаковой частотой как у мальчиков, так и у девочек. За почти сто лет, прошедших с момента первого описания, подробно изучено около 400 случаев данной патологии.
Аномалии головы и кожи
Синдром Корнелии де Ланге можно заподозрить с первых минут жизни младенца. Первичные симптомы:
1. Малый вес новорожденного (примерно 2/3 от нормы).
2. Аномалии черепа:
— микроцефалия (череп примерно на 10 % меньше нормы (у 98 % больных);
— брахицефалия (увеличение поперечного размера (ширины) черепа по сравнению с его продольными размерами);
— уменьшение мозговой части головы.
Больше чем у половины новорожденных наблюдается повышенная волосистость спины, а иногда и всего тела. Кожа у примерно 2/3 младенцев имеет синюшный рисунок с хорошо просматриваемыми сосудами (мраморность кожи), но этот симптом не является определяющим в диагностике данного заболевания.
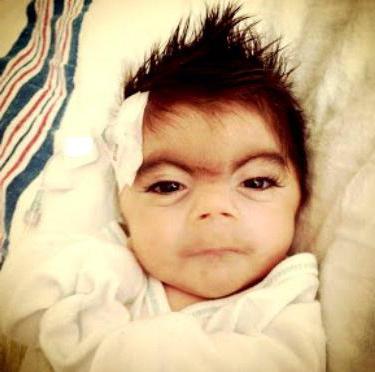
Аномалии лица
Дефекты на лице новорожденного являются наиболее яркими критериями, чтобы диагностировать синдром Корнелии Де Ланге. Фото малыша выше наглядно это показывает. К отклонениям от общепринятых норм относятся:
— четко очерченные, как нарисованные брови, сходящиеся на переносице (99 % случаев);
— красивые, длинные ресницы, часто загнутые назад (99 %);
— маленький носик, на котором ноздри выпячиваются вперед (88 %);
— широкая и запавшая переносица (88 %);
— рот с опущенными уголками (94%);
— нестандартно большое расстояние между носом и губами;
— низко расположенные уши;
— недоразвитие (гипоплазия) нижней челюсти (84 %);
— слишком низко расположена на лбу и/или затылке граница волосяного покрова (94 %).
Вышеперечисленные отклонения на лице новорожденного могут присутствовать все, а могут только некоторые.
Аномалии внутренних органов
Гипоплазия (недоразвитие) и патологические нарушения в формировании внутренних органов представляют наибольшую опасность для жизни детей, у которых выявлен синдром Корнелии де Ланге. Диагностика включает МРТ, рентген, УЗИ, риноскопию, цитогенетические анализы и другие современные методы.
При данном заболевании может наблюдаться:
— атрезия хоан, а проще говоря, непроходимость полости носа (эта патология бывает совершенно не связана с синдромом де Ланге и легко диагностируется по тому, как младенец дышит либо с помощью зонда);
— дефекты в строении сердца (пороки сосудов, клапанов, перегородок);
— пороки ЖКТ (слепая кишка подвижна, некоторые другие);
— пороки мочеполовой системы (примерно у 50 % больных);
— кисты на почках, гидронефроз;
— патологии в мозговой ткани (дисплазия извилин, аплазия мозолистого тела и другие);
— слишком высокое либо с расщелиной нёбо;
— крипторхизм.
Опять-таки необязательно, чтобы у одного больного ребенка присутствовали все вышеперечисленные дефекты внутренних органов.
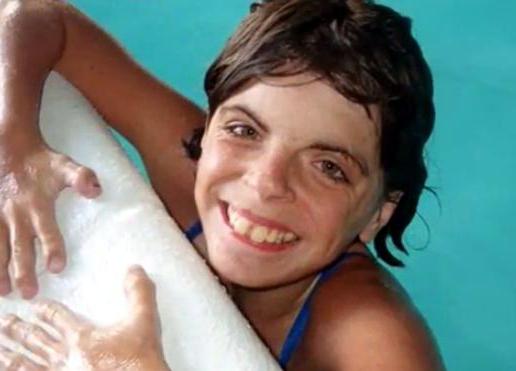
Аномалии костно-мышечного аппарата
У новорожденных могут быть дефекты конечностей, позвоночника, грудной клетки, по которым также диагностируется синдром Корнелии Де Ланге (фото людей с этим заболеванием представлены в статье).
Некоторые дефекты видны сразу. Это:
— отсутствие одного или нескольких пальцев на руках;
— сросшиеся пальцы (чаще встречается на ногах);
— деформированные позвоночник и/или грудная клетка.
С взрослением ребенка становятся довольно значительными следующие отклонения:
— отставание в росте (в некоторых случаях нанизм);
— недоразвитие, укорочение конечностей;
— маленькие кисти рук и стоп;
— слишком короткая шея;
— ограничение в способности локтевых суставов сгибаться-разгибаться (контрактура).
Неврологические отклонения и патологии органов чувств
К сожалению, есть множество других проблем у детей, которым поставлен диагноз «синдром Корнелии де Ланге». Симптомы заболевания, связанные с неврологическим состоянием ребенка, могут быть такими:
— новорожденные вяло сосут, часто срыгивают;
— малая подвижность и двигательная активность;
— гипотония мышц (мышечный тонус понижен, нет силы в руках и ногах);
— периодическое возникновение судорог.
Дети с синдромом де Ланге имеют проблемы со слухом, зрением и речью. Многие из них плохо говорят или почти не говорят в любом возрасте. Родители отмечают, что в большинстве случаев малыши свои желания выражают жестами. Со зрением у них бывают такие проблемы:
— косоглазие;
— близорукость;
— астигматизм;
— атрофия зрительного нерва.

Интеллектуальное развитие
Синдром Корнелии де Ланге кроме всех прочих осложнений со здоровьем вызывает умственную отсталость, которая отмечается практически у каждого больного ребенка, причем в 80 % случаев диагностируется имбецильность либо дебильность. Однако есть дети с синдромом де Ланге, которые посещают обычные дошкольные и школьные учреждения. Это зависит от того, в какой из двух форм диагностировано заболевание. Первая называется классической, при которой наблюдаются многие отклонения в формировании и работе внутренних органов, внешние аномалии и ярко выраженная задержка умственного развития. Вторая форма условно называется смазанной. При ней наблюдаются те или иные внешние отклонения, есть некоторые проблемы с внутренними органами, но интеллектуальное развитие имеет пограничную задержку.
Как отмечают родители, дети с синдромом де Ланге в любом возрасте не просятся в туалет, часто подвержены раздражению, любят выполнять различные несвойственные здоровым детям действия: постоянно рвать бумагу, есть ее, ломать все, что попадает им в руки, все время двигаться по кругу. Такие действия детей как бы их успокаивают.
Причины заболевания
Прошло практически сто лет с момента, когда впервые был описан синдром Корнелии де Ланге. Причины заболевания за это время удалось выяснить. Ими являются мутации в генах. Наибольшее число случаев зафиксировано при мутациях в 5-й хромосоме, точнее в гене NIPBL, находящемся в ее плече «р». Небольшой процент возникновения синдрома де Ланге зафиксирован у людей с мутацией 1А-белка хромосом (называется ген SMC1A), и один случай отмечен с мутацией также в белке хромосом, называемом геном SMC3. Однако причины, вызывающие эти генные мутации, пока находятся в стадии теорий и предположений.
Есть мнение, что их могут вызывать инфекционные заболевания беременной, особенно в первом триместре, некоторые лекарственные препараты, преклонный возраст отца ребенка либо возраст рожениц старше 35 лет, а также браки между родственниками.
Но ни одна из перечисленных причин не является бесспорной и 100 % вызывающей генетические изменения.
Еще прорабатывается гипотеза, что синдром Корнелии де Ланге передается по наследству, однако больше чем у половины обследованных пациентов это заболевание возникло спорадически, впервые в роду.
Синдром Корнелии де Ланге: прогноз
Считается, что данное заболевание очень редкое. Единых данных, с какой частотой оно встречается, нет. Одни источники утверждают, что заболевает один ребенок на 10 000 новорожденных, другие, что один на 100 000, третьи называют различные цифры в этом диапазоне. Если вдуматься, это не так уж и мало. Согласно статистике ежедневно на земле рождается около 370 тысяч младенцев. То есть, если взять даже самые низкие показатели, каждый день рождается примерно 4 человека, у которых диагностируется синдром Корнелии де Ланге.
Сколько живут такие люди, зависит от многих факторов, определяющими из которых являются степень аномалий внутренних органов, своевременное их выявление и качество оказанных медицинских воздействий. Если у ребенка патологии внутренних органов, несовместимые с жизнью, он умирает в первый месяц после рождения. Если же аномалии внутренних органов незначительны или ребенку было вовремя произведено хирургическое вмешательство, срок его жизни может быть достаточно долгим. Осложняет прогноз то обстоятельство, что организм больных с синдромом де Ланге не способен оказывать стойкое сопротивление обычным болезням, например вирусным, и труднее с ними борется.

Лечение
Большинство авторов и источников утверждают, что не существует возможности вылечить ребенка, у которого диагностирован синдром Корнелии де Ланге. Лечение сводится к хирургическим операциям (если есть показания), приему витаминов, ноотропных средств (воздействуют на функции мозга), анаболиков, проведении седативной терапии. Однако в наше время высочайших технологий можно если не полностью победить недуг, то значительно снизить его проявление. Для этого нужны вера в успех, нечеловеческое терпение и деньги, потому что лечение дорогое. Вот контакты клиник и центров, где берутся помочь:
1. Киев. Научно-методический центр «Истина», расположен на улице Вильямса, строение № 4. Телефоны: +38-044-467-63-89, +38-095-068-30-74. Здесь работают по методу Ульяны Лущик, есть много положительных отзывов.
2. Москва, Солнцево. Научно-практический центр, расположенный на улице Авиаторов, строение № 38. Телефоны: +7-495-934-17-53, +7-495-934-27-10, +7-495-934-14-39. Есть много отзывов, что детям с синдромом де Ланге после курсов лечения в этом центре становится гораздо лучше.
3. Израиль. Центр биокоррекции им. Васильева (лечение стоит от 10 тысяч у. е.). Телефон: 972-352-333-89.
У детей с диагнозом «синдром Корнелии де Ланге» продолжительность жизни во многом зависит от самоотверженной заботы о них родных людей, ведь заниматься с такими больными нужно практически каждую минуту. Очень часто достигнутые положительные результаты лечения уменьшаются или сводятся к нулю, если прекращают лечение или просто потому что происходит рецидив.
По отзывам родителей, положительные результаты в лечении их детей дают кинезотерапия, специальные реабилитационные программы плавания с дельфинами в дельфинарии, биоритмокоррекция, ароматерапия, музыкальные занятия, светотерапия.
Профилактика
Трудно говорить о профилактических мероприятиях заболевания, причины возникновения которого никому не известны. Учитывая установленные факторы, способные вызвать мутации на генетическом уровне, можно посоветовать:
— не допускать зачатия детей от кровных родственников;
— с осторожностью относиться к поздним материнству и отцовству;
— в период беременности, особенно в первые месяцы, принимать все меры, чтобы избежать вирусных инфекций, а в случае заболевания принимать лекарства только после консультации с врачом.
На основе проведенных исследований пациентов с синдромом Корнелии де Ланге врачи склонны считать, что в одной семье повторно может родиться больной ребенок в 2 % случаев, а в тех семьях, где у отца или у матери присутствуют некоторые симптомы синдрома де Ланге, больной ребенок может родиться в 25 % случаев. В связи с этим все женщины из группы риска должны проходить углубленную пренатальную диагностику, в частности проверять наличие в сыворотке крови белка РАРРА. Если он отсутствует, есть очень высокая вероятность родить ребенка с синдромом Корнелии де Ланге.
Источник