Код мкб 10 миопатия

Рубрика МКБ-10: G72.9
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G70-G73 Болезни нервно-мышечного синапса и мышц / G72 Другие миопатии
Определение и общие сведения[править]
Миопатии — группа наследственных заболеваний, проявляющихся мышечной слабостью и атрофией мышц. Прогрессирующие миопатии называют также миодистрофиями. Гистологически выявляются снижение числа мышечных волокон и вариабельность размеров оставшихся. При амиотрофиях (атрофиях мышц при поражениях двигательных нервов или мотонейронов) патологоанатомическая картина иная. Существует несколько типов миопатий (см. табл. 16.2).
Тяжесть и скорость прогрессирования заболевания зависит от типа миопатии и индивидуальных особенностей. Наиболее тяжело протекает миопатия Дюшенна, при которой большинство больных не доживают до 20 лет. При других миопатиях больные достигают зрелого возраста. Существуют легкие формы миопатий, поражающие лишь определенные группы мышц и практически не ограничивающие жизнедеятельность.
Этиология и патогенез[править]
Патогенез большинства миопатий остается неизвестным. Недавно показано, что в основе этих заболеваний может лежать дефект мембраны мышечных клеток. Миопатии Дюшенна и Беккера связаны с делецией в локусе Xp21 (на коротком плече X-хромосомы). При миопатии Дюшенна в мембране мышечных клеток отсутствует продукт данного гена — дистрофин, а при более доброкачественной миопатии Беккера наблюдается снижение содержания дистрофина либо выявляется дистрофин с аномальным молекулярным весом. Патогенез других миопатий менее изучен.
Клинические проявления[править]
Миопатия неуточненная: Диагностика[править]
1. Выявление в анамнезе и при осмотре у детей и подростков прогрессирующей слабости и похудания мышц позволяет заподозрить миопатию. Однако по клиническим данным невозможно полностью отличить миопатию от амиотрофии. Различные типы миопатий отличаются друг от друга преимущественным поражением тех или иных мышечных групп, скоростью прогрессирования и другими клиническими проявлениями (см. табл. 16.2). В табл. 16.3 приведены признаки, позволяющие отличить амиотрофии от миопатий.
2. Биопсия мышц подтверждает диагноз миопатии и позволяет отличить тяжелые прогрессирующие миопатии от доброкачественных непрогрессирующих (например, от болезни центрального стержня или немалиновой миопатии). Биоптат берут из пораженной, но не наиболее ослабленной мышцы. Чаще всего при миопатиях исследуют дельтовидную и икроножную мышцы, хотя целесообразней проводить биопсию прямой мышцы живота — это не нарушает двигательную активность больного и не приводит к дальнейшему нарастанию слабости вследствие бездействия.
3. Исследование ДНК лейкоцитов с помощью ПЦР выявляет дефект у 70% больных с миопатией Дюшенна и Беккера. Данным методом можно также исследовать культуру ткани плода. Исследование дистрофина в мышечном биоптате позволяет отличить миопатии Дюшенна и Беккера друг от друга и от других миопатий, при которых содержание и структура дистрофина не меняются. Хотя сочетание этих двух методов значительно повышает точность диагноза, в большинстве случаев биопсию не проводят, если при исследовании ДНК лейкоцитов обнаружена характерная делеция.
Дифференциальный диагноз[править]
Миопатия неуточненная: Лечение[править]
1. В легких случаях лечение, особенно на ранней стадии, не требуется; достаточно повторных (каждые 6—12 мес) исследований двигательной функции.
2. В тяжелых случаях, особенно при миопатии Дюшенна, необходимо разъяснить родственникам суть и прогноз заболевания и заручиться их поддержкой.
а. Скрупулезное выполнение назначений врача способно продлить период относительной независимости больного, и семья должна это хорошо понимать. По мере прогрессирования заболевания часто наступает разочарование, однако и в этой ситуации важно поддерживать надежду на стабилизацию процесса и настаивать на продолжении физических упражнений.
б. Поскольку большую часть больных с миопатиями составляют дети, необходимо предусмотреть возможность получения образования и общения со сверстниками. Пока ребенок может без особого труда подниматься по лестнице, он должен посещать обычную школу. В то же время при миопатии Дюшенна интеллект часто снижается (IQ у многих больных составляет менее 90). В таких случаях детей лучше сразу направлять в специальную школу.
в. Цель лечения состоит не только в том, чтобы сохранить способность к самостоятельному передвижению. Болезнь не должна заполнять все существование больного — следует стремиться к тому, чтобы он как можно дольше вел нормальный образ жизни.
3. Преднизон (0,75 мг/кг/сут) способствует увеличению мышечной силы при миопатии Дюшенна. Однако его польза не перевешивает риск побочных эффектов, и поэтому его обычно не назначают. Исключение составляют случаи, когда в результате пневмонии или ателектаза развиваются острые дыхательные расстройства. Чтобы избежать прибавки в весе, во время лечения назначают диету с низким содержанием жиров и соли. При возможном контакте с вирусом varicella-zoster больным, принимающим кортикостероиды, назначают иммуноглобулин против вируса varicella-zoster.
4. ЛФК
а. Тренировки начинают как можно раньше. Больного и его родных следует обучить комплексам упражнений. ЛФК более эффективна, если ее начинают до появления контрактур и деформаций.
б. Цель ЛФК — обеспечить функционирование опорных суставов и предотвратить контрактуры. Так как сгибатели поражаются в несколько меньшей степени, чем разгибатели, контрактуры (в тазобедренных, локтевых и коленных суставах) обычно носят сгибательный характер.
в. Для профилактики контрактур, затрудняющих передвижение и усложняющих уход за больным, необходимы упражнения на объем движений, коррекция положения тела в кровати и кресле, частая смена позы, раннее применение шин.
г. Хотя нет доказательств, что ЛФК замедляет прогрессирование болезни, она тем не менее позволяет отсрочить на несколько лет обездвиженность.
5. Лечение дыхательных расстройств
а. В тяжелых случаях генерализованной миопатии и при поражении мышц гортани и глотки возникают нарушения глотания и дыхания. Дыхательная недостаточность обычно нарастает постепенно и становится явной лишь на поздней стадии.
б. При исследовании функции внешнего дыхания часто выявляются нарушения даже в отсутствие явных дыхательных расстройств. Целесообразно тренировать диафрагмальное дыхание (надувать игрушки или играть на духовых инструментах). Необходимы и специальные дыхательные упражнения под контролем инструктора.
в. На поздней стадии приходится прибегать к ИВЛ и постуральному дренажу.
г. Гиперкапния в отсутствие пневмонии — плохой прогностический признак, поскольку 80% больных с тяжелой миопатией погибает от дыхательной недостаточности. К интубации трахеи при миопатиях прибегают редко.
6. Поддержание подвижности
а. Уменьшение избыточного веса улучшает двигательные возможности и предотвращает гиповентиляцию.
б. Целесообразны ежедневные пешие прогулки продолжительностью не менее 3 ч. Если больной не может ходить, то ему рекомендуют стоять в общей сложности 3 ч в сутки (по 30 мин каждые 3—4 ч).
в. По мере прогрессирования заболевания при ходьбе можно использовать костыли и другие ортопедические приспособления. К передвижению в кресле-каталке переходят как можно позже.
г. При острых сопутствующих заболеваниях слабость может усиливаться, однако постельный режим обычно противопоказан, так как он приводит к еще большему нарастанию мышечной слабости и некоторые больные больше уже никогда не встают.
Профилактика[править]
Профилактика и медико-генетическое консультирование. Если тип наследования заболевания известен, можно предсказать вероятность его развития у ребенка. Больным с миопатиями следует обязательно сообщать степень риска для их будущего потомства.
а. Сцепленные с полом рецессивные заболевания (миопатии Дюшенна и Беккера)
1) Наследование. Вероятность рождения у носителя патологического гена больного сына или дочери-носительницы составляет 50%. Однако примерно у трети больных заболевание в семейном анамнезе отсутствует.
2) Выявление носителей. Примерно у половины носителей в сыворотке увеличена активность КФК. Поскольку активность КФК может колебаться, исследование нужно проводить по меньшей мере 3 раза с интервалом в 10 сут. Накануне взятия крови ограничивают нагрузки, так как они приводят к повышению КФК. Нормальные результаты не исключают возможность носительства. В 60—65% семей с миопатиями Дюшенна или Беккера выявляются характерные мутации в виде интрагенных делеций гена дистрофина. Носительниц в семье больного мальчика можно выявить путем обнаружения делеции в одном из двух аллелей, кодирующих дистрофин. В других случаях используют метод, основанный на анализе полиморфизма длин рестрикционных фрагментов. Точность выявления носительства у девочек достигает 85—90%.
3) Профилактика. Носители могут предотвратить рождение больного ребенка с помощью нескольких способов.
а) Добровольная стерилизация или контрацепция.
б) Пренатальные определение пола и диагностика позволяют произвести аборт по медицинским показаниям.
4) Спонтанная мутация лежит в основе трети случаев миопатии Дюшенна. Чем больше здоровых детей родилось в семье до появления больного ребенка, тем меньше вероятность, что мать является носителем. В этом случае вероятность рождения у нее еще одного больного ребенка невелика.
б. Аутосомно-доминантные заболевания (плече-лопаточно-лицевая миопатия, поздняя дистальная миопатия)
1) Наследование. В типичном случае плече-лопаточно-лицевая дистрофия передается по аутосомно-доминантному типу, однако существуют другие формы с аналогичной клинической картиной, которые наследуются по аутосомно-рецессивному или сцепленному с полом рецессивному механизму. Тяжесть заболевания варьирует. При аутосомно-доминантном типе наследования вероятность передачи заболевания потомству составляет 50%.
2) Носительства не бывает.
3) Профилактика возможна с помощью контрацепции. Если болен мужчина, то для появления здорового потомства можно прибегнуть к искусственному оплодотворению.
в. Аутосомно-рецессивные заболевания (тазо-плечевая миопатия)
1) Наследование. Если оба родителя являются носителями, то один из четырех детей будет болен, два из четырех будут носителями и только один будет полностью здоров.
2) Профилактика: контрацепция и воздержание от близкородственных браков.
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Bieber, F. R., Hoffman, E. P., and Amos, J. A. Dystrophin analysis in Duchenne muscular dystrophy: Use in fetal diagnosis and in genetic counseling. Am. J. Hum. Genet. 45:362, 1989.
2. Fenichel, G. M., et al. Long term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 41:1874, 1991.
3. Hardiman, O., et al. Neuropathic findings in oculopharyngeal muscular dystrophy: A report of seven cases and a review of the literature. Arch. Neurol. 50(5):481, 1993.
4. Harris, S. E., and Cherry, D. B. Childhood progressive muscular dystrophy and the role of physical therapy. Phys. Ther. 54:4, 1974.
5. Hook, R., Anderson, E. F., and Noto, P. Anesthetic management of a parturient with myotonia atrophica. Anesthesiology 43:689, 1975.
6. Iannaccone, S. T. Current status of Duchenne muscular dystrophy. Pediatr. Clin. North Am. 39(4):879, 1992.
7. Love, D. R., et al. Dystrophin and dystrophin-related proteins: A review of protein and RNA studies. Neuromuscul. Disord. 3(1):5, 1993.
8. Miller, G., and Wessel, H. B. Diagnosis of dystrophinopathies: Review for the clinician. Pediatr. Neurol. 9(1):3, 1993.
9. Sarnat, H. B., O’Connor, T., and Byrne, P. A. Clinical effects of myotonic dystrophy on pregnancy and the neonate. Arch. Neurol. 33:459, 1976.
10. Wessel, H. B. Dystrophin: A clinical perspective. Pediatr. Neurol. 6(1):3, 1990.
11. Zellweger, M. D., and Ionasecu, V. Myotonic dystrophy and its differential diagnosis. Acta Neurol. Scand. (Suppl. 55) 49:1, 1973.
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Патогенез
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Миопатии.
Миопатии
Описание
Миопатии. Группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Дополнительные факты
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Особенности отдельных форм миопатии.
Ювенильная миопатия Эрба наследуется аутосомно. Рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече. Лопаточно — лицевая миопатия Ландузи — Дежерина имеет аутосомно — доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия. Аутосомно — доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно. Доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
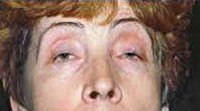
Миопатии
Симптомы
Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Патогенез
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр. ), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и тд ), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гипотиреоза, гипертиреоза, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и тд формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные — возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при синдроме Шегрена, СКВ, склеродермии и тд коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов — уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и тд ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник