Кариотип человека с синдромом патау

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 мая 2019;
проверки требуют 7 правок.
Синдром Пата́у (трисомия 13) — генетическое заболевание человека, которое характеризуется возникновением геномной мутации, а именно трисомией по 13-й хромосоме.
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657 году. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь. Синдром Патау также был описан для племён с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе.
Этиология и эпидемиология
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трёх из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель). Риск возникновения этого синдрома у потомства увеличивается с возрастом матери, достигая пика в среднем к 31 году[2].
Проявления заболевания
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
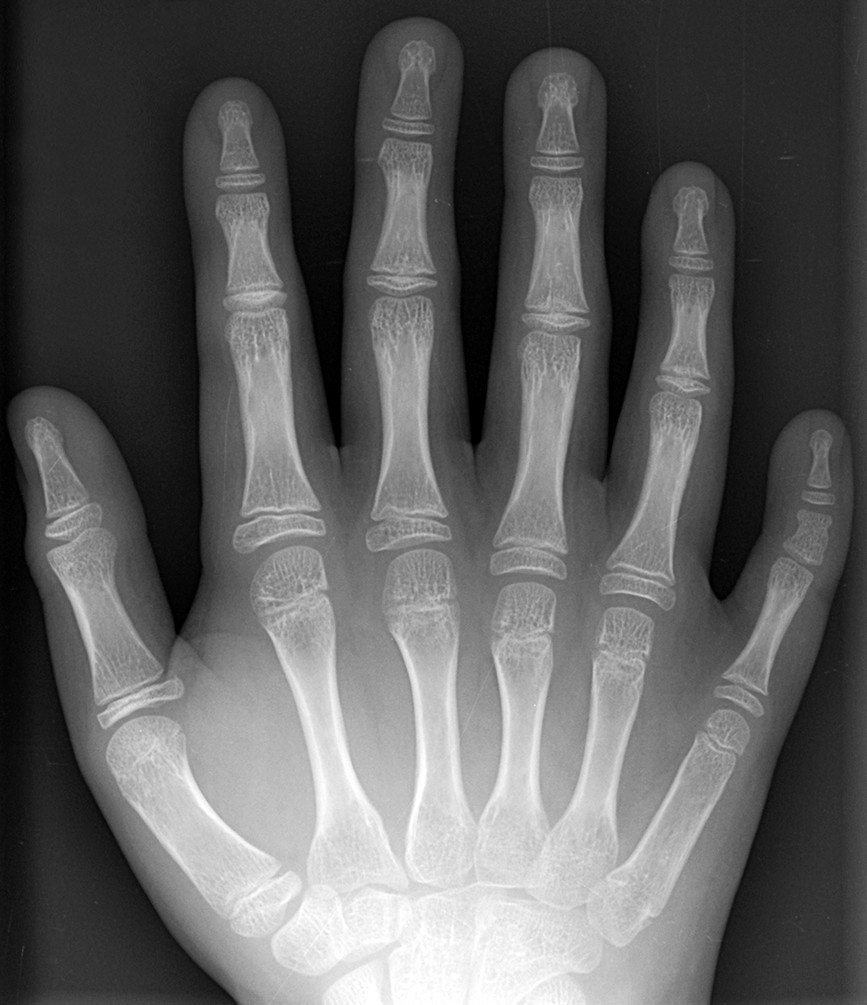
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
См. также
- Хромосомные заболевания
- Синдром Дауна
- Синдром Эдвардса
Примечания
Ссылки
- https://web.archive.org/web/20070929094344/https://schools.keldysh.ru/school1413/pro_2005/z/hrbol.htm
- https://www.medkurs.ru/lecture2k/genetics/gl19/4288.html
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
Количество хромосомных заболеваний огромно и одним из них является синдром Патау. Кариотип пациента с подобным диагнозом изменен, что отображается на работе всего организма. Патология сказывается на строении скелета, работе нервной, выделительной, репродуктивной и сердечно-сосудистой систем.
Многие родители сегодня интересуются любой дополнительной информацией о данной патологии. Что представляет собой синдром Патау? Кариотип и фенотипическая характеристика, методы диагностики и лечения, симптомы и причины — все это интересные моменты. Такие вопросы стоит изучить более подробно.
Синдром Патау: кариотип и общая информация

Для начала стоит разобраться с общими данными. Что собой представляет и как развивается синдром Патау? Хромосомы при подобной патологии не расходятся в процессе образования гамет или зиготы, что приводит к массе нарушений в процессе эмбрионального развития.
Синдромом Патау называют врожденное заболевание, которое связано с трисомией 13-й пары хромосом — ребенок получает лишнюю хромосому. Формула кариотипа в данном случае может выглядеть следующим образом: 47, XX, 13 + (для девочек), 47, XY, 13 + (для мальчиков). Это крайне опасное и тяжелое заболевание, которое сопровождается формированием множественных пороков развития — часто они не совместимы с жизнью, поэтому ребенок гибнет еще в утробе матери.
Краткая историческая справка
Впервые симптомы данной патологии были описаны в 1657 году в работах датского ученого Эразмуса Бартолина. Тем не менее хромосомная природа заболевания была доказана доктором Клаусом Патау в 1960 году (в его честь и был назван синдром).
Случаи подобных врожденных патологий были описаны учеными, которые занимались исследованием племен Тихоокеанских островов. Считается, что учащенные хромосомных мутаций в этой географической области были вызваны радиационным поражением, появившимся после испытаний ядерного оружия.
Причины развития заболевания
Как уже упоминалось, синдром Патау характеризуется трисомией тринадцатой пары хромосом. При этом в большинстве случаев (согласно статистике, в 80 %) имеет место нерасхождение тринадцатой хромосомы именно в процессе мейоза. Чаще всего ребенок получает полную пару хромосом от матери. Иногда имеет место робертсоновская транслокация, при которой уже зародыш получает дополнительную копию генов.
Доказано, что генетический сбой может произойти во время формирования гаметы или позднее — при образовании зиготы.
На сегодняшний день причины появления мутации не установлены — считается, что она возникает абсолютно случайно. Также неизвестна роль инфекций, вредных привычек, соматических заболеваний матери в процессе развития данной патологии у плода.
Виды патологии
На сегодняшний день выделяют несколько форм данного заболевания.
- Простая форма. В данном случае нарушения происходят на ранних этапах развития зародыша. При этом каждая клетка организма содержит в себе лишнюю хромосому в тринадцатой паре.
- Мозаичная форма. Подобные изменения происходят уже на более поздних этапах развития плода. При этом некоторые органы состоят лишь из здоровых клеток, в то время в других тканях и органах содержатся патологически измененные клетки. При данной форме заболевания симптомы могут быть менее выраженными.
Существуют ли факторы риска? Что может спровоцировать появление мутации?
Синдром Патау развивается спонтанно и предупредить его, увы, невозможно. Тем не менее учеными выделено несколько неблагоприятных факторов.
- Считается, что риск появления подобной патологии повышается в том случае, если мать проживает в районе с плохой экологической ситуацией.
- Как уже упоминалось, определенную роль в появлении спонтанных хромосомных мутаций играет радиационное облучение.
- Замечено также, что вероятность появления различных хромосомных и генетических мутаций увеличивается в случае поздней беременности (мать старше 45 лет).
- К факторам риска относят наличие наследственных заболеваний в предыдущих поколениях родителей.
- Доказано, что случаи генетических и хромосомных мутаций учащаются если речь идет о браках между близкими родственниками.
Симптомы патологии при беременности

Как уже упоминалось, формула кариотипа при синдроме Патау изменена, но обнаружить это без специальных тестов невозможно. Тем не менее часто такая беременность сопровождается многоводием. Согласно статистике, время вынашивания плода также сокращается — в среднем составляет около 38,5 недель. Стоит отметить, что при подобной патологии беременность часто прерывается. Существует большой риск мертворождения.
Синдром Патау: фото и отличительные признаки
Дети с подобной мутацией имеют ряд особых фенотипических характеристик. Для начала стоит сказать, что малыши рождают в срок, но с маленьким весом — имеет место пренатальная гипотрофия (вес доношенного ребенка с подобным диагнозом редко превышает 2500 г). Сам процесс родов также часто сопряжен с осложнениями, в частности асфиксией новорожденного.
Ребенок с хромосомной мутацией имеет ряд врожденных деформаций мозговой и лицевой части черепа. Имеет место микроцефалия — окружность головы малыша значительно меньше нормы. У детей с синдромом Патау лоб часто низкий и скощенный, переносица плоска и запавшая, а глазные щели узкие. Ушные раковины, как правило, деформированы и располагаются низко.
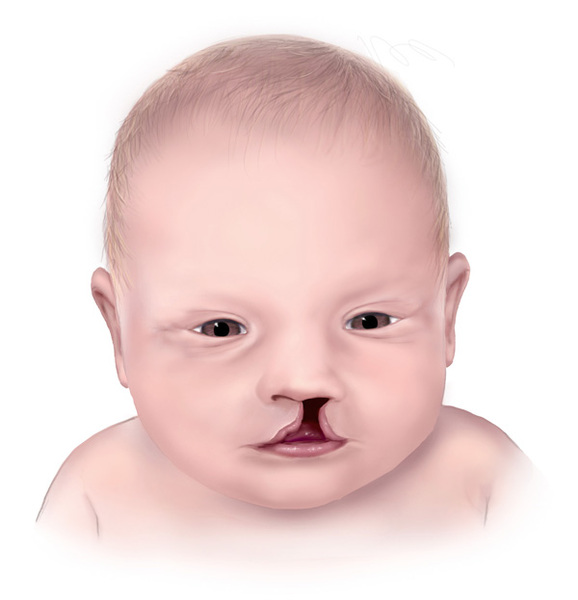
Типичным симптомом является и наличие двухсторонних расщелин лица, в частности, так называемой волчьей пасти (патология сопровождается расщеплением тканей мягкого и твердого неба, причем носовая и ротовая полости сообщены между собой), а также «заячьей губы» (расщелина губы). Врач может заподозрить наличие данной хромосомной мутации уже в первые часты после рождения ребенка.
Патология со стороны других систем органов
Какими еще нарушениями сопровождается синдром Патау? Признаки лицевых деформаций являются отнюдь не единственными. Недуг затрагивает работу практически всех систем органов, вызывая тяжелые врожденные пороки.
- У многих детей наблюдаются нарушения со стороны центральной нервной системы и зрительных анализаторов. Возможна гидроцефалия, гилопрохэнцефалия, дисгинезия мозолистого тела, а также образование спинномозговых грыж. К возможным осложнениям относят глухоту, врожденные формы катаракты, дисплазию сетчатки, уменьшение количества аксонов в структуре зрительного нерва, что ведет к серьезным нарушениям зрения.
- Возможны врожденные пороки сердца, включая открытый аортальный проток, коарктацию аорты, дефекты межжелудочковой и межпредсердной перегородки.
- Многие дети рождаются с пороками развития почек. Например, у пациентов диагностируются гидронефрозы, поликистозы, а также патология, носящая название «подковообразной почки».
- Возможны аномальные изменения со стороны пищеварительного тракта. К их списку относят образование дивертикулов Меккеля, кистозные новообразования в тканях поджелудочной железы, незавершенный поворот кишечника.
- Мутация затрагивает и органы репродуктивной системы. У мальчиков возможна задержка опускания яичек в мошонку (крипторхим), а также смещение наружного отверстия уретры. У новорожденных девочек нередко наблюдается гипертрофия половых губ и клитора, формирование двурогой матки или даже образование двух обособленных маток и влагалищ.
- Патология затрагивает не только кости черепа, но и другие структуры скелета. Например, у детей с синдромом Патау часто наблюдается полидактилия (появление дополнительных пальцев на стопах и кистях) и синдактилия (сращение пальцев).
- Все дети с данным заболеванием страдают от выраженных форм физической и умственной отсталости.
Как можно увидеть, данная хромосомная патология имеет опасные и тяжелые последствия, поэтому прогнозы для детей неблагоприятны.
Диагностические мероприятия
Можно ли во время внутриутробного развития выявить такое заболевание, как синдром Патау? Диагностика на этом этапе, безусловно, возможна. Как уже упоминалось, патология сопровождается возникновением тех или иных симптомов еще во время беременности.
Как правило, подозрения на наличие подобной мутации появляются во время УЗИ. Специалист может отметить увеличение объема околоплодных вод, а также наличие нарушений в анатомическом строении скелета, отсутствие у плода волос, низкую массу тела плода и т. д.
В дальнейшем проводятся генетические исследования наследственного материала, ведь, как известно, кариотип больного с синдромом Патау отличается от нормального. Информативными являются следующие тесты.
- Амниоцентез – забор и дальнейший анализ околоплодных вод.
- Кордоцентез — суть методики заключается в получении образцов пуповинной крови с дальнейшим ее лабораторным исследованием.
- Биопсия ворсинок хориона — процедура, которая предусматривает забор ворсинок с помощью специального зонда. Этот тест назначают женщинам при подозрении на синдром Дауна, Патау и прочие хромосомные мутации.
Стоит отметить, что подобные исследования сопряжены с риском (например, существует вероятность потери ребенка). После рождения малыша диагноз подтверждается после осмотра неонатолога или педиатра.
Существует ли эффективное лечение?
Вы уже знаете о том, что представляет собой синдром Патау. Кариотип ребенка с таким диагнозом изменен, что сказывается на структуре и функционировании всего организма. К сожалению, возможности для проведения терапии очень ограничены. Иногда проводится хирургическое вмешательство, которое позволяет устранить анатомические аномалии (например, расщелины лица).
Ребенок должен постоянно наблюдаться у детского кардиолога, невролога, офтальмолога, уролога, ЛОРа, ортопеда, генетика и прочих специалистов. Малышу нужно обеспечить хороший уход и полноценное питание. В зависимости от наличия тех или иных нарушений проводится симптоматическая терапия.
Прогнозы для маленьких пациентов

На сегодняшний день крайне опасным считается синдром Патау. Кариотип ребенка изменить невозможно и прогнозы для детей крайне неблагоприятны. Очень часто беременность заканчивается выкидышем.
Если малыш все же рождается, то, согласно статистике, редко доживает до одного года. Лишь в некоторых развитых странах у врачей и родителей есть возможность продлить жизнь ребенка до 4-5 лет. К сожалению, на сегодняшний день медицина не может предложить эффективные методы профилактики или лечения.
Источник

Синдром Патау – генетически детерминированный недуг, развивающийся у лиц, которые имеют лишнюю 13 хромосому. Проявляется данная хромосомная аномалия признаками поражения нервной системы, зрительного анализатора, костей, мышц, сердца, органов мочевыделительной системы. Характерен внешний вид больных. У них уменьшен размер черепа и глаз, головной мозг не разделен на полушария, имеются дополнительные пальцы, расщелины губы и неба, грыжа пупочного канатика, дефекты в структуре сердца и крупных сосудов. Подобные аномалии обуславливают антенатальную смерть плода и малую продолжительность жизни больных детей.
Впервые клинические проявления патологии описали медики еще в XVII веке. Спустя несколько столетий ученый Патау определил, что заболевание связано с появлением дополнительной 13 хромосомы в кариотипе человека. Благодаря этому открытию синдром получил свое название. Современные ученые установили, что одной из причин патологии является ионизирующее излучение.

выживший ребенок с синдромом Патау
Диагноз синдрома Патау устанавливают по результатам пренатального скрининга или кариотипирования новорожденного. Среди всех генетических мутаций этот недуг встречается относительно часто и уступает по распространенности только синдрому Дауна. Согласно статистике, расовые, географические и половые факторы не влияют на частоту развития патологии. Постнатальная диагностика довольно проста: серьезные деформации частей тела видны невооруженным глазом.
Поскольку синдром Патау является генетическим заболеванием, специальные лечебные методики, предназначенные для его устранения, не разработаны. Больным показано консервативное лечение, улучшающее их общее состояние, а также оперативное вмешательство, ликвидирующее врожденные дефекты.
Причины
Синдром Патау развивается в случае появления в генотипе плода дополнительной 13 хромосомы. Современные ученые до сих пор не могут объяснить, из-за чего же так происходит. Возможно всему виной гормональный сбой, сопровождающий процесс образования половых клеток или формирования зиготы. Больной ребенок может появиться на свет в семье, где родители и ближайшие родственники являются абсолютно здоровыми.

Факторы, способствующие развитию патологии:
- Беременность после 40 лет,
- Острые инфекционные патологии матери,
- Употребление алкоголя или курение во время беременности,
- Неблагоприятная экология,
- Интимная связь с близкими родственниками,
- Отягощенный наследственный анамнез,
- Облучение,
- Воздействие химикатов, лекарств и прочих токсинов,
- Заболевания органов репродуктивной системы,
- Эндокринопатии.
Вышеперечисленные факторы лишь повышают риск развития данной мутации, а не являются ее непосредственными причинами.
Чтобы жить, расти и развиваться, человеку необходимы две копии каждой хромосомы. Появление третьей, добавочной, хромосомы смертельно опасно для плода. Синдром Патау – одна из тех трисомий, при которых возможно живорождение.
Основные формы синдрома Патау:
- Цитогенетическая – появление дополнительной хромосомы связано с тем, что они не расходятся в процессе мейотического деления клеток. Эта форма встречается чаще всего и отличается очень тяжелым течением.
- Простая – на ранних этапах развития плода каждая клетка уже имеет лишнюю хромосому по причине обширной мутации или имеющихся отклонений в яйцеклетке до оплодотворения.
- Мозаичная – на поздних этапах развития плода одни области имеют здоровые клетки, а другие патологичные, что приводит к аномалии отдельных органов ребенка.
Подобные патологические изменения происходят в организме плода во время эмбриогенеза.
Симптомы
Первые клинические признаки патологии обнаруживаются уже при беременности. Срок ее сокращается до 38 недель, а течение осложняется многоводием. Врожденные аномалии, формирующиеся при синдроме Патау, обычно приводят к гибели плода внутри утробы матери. Выкидыши и мертворождение — самый частый исход патологии.

синдром Патау различной тяжети
Нередко в процессе родов младенцы задыхаются. Они рождаются с очень малой массой тела, так называемой пренатальной гипотрофией. Отличительные признаки заболевания у больных детей — симптомы поражения костно-суставного и мышечного аппарата, ЦНС, органа зрения, внутренних органов.
- Особенности внешнего вида: килевидная деформация черепа, микроцефалия, атипичный размер и форма лба, близко посаженные орбиты, сужение глазной щели, широкий нос, низкие ушные раковины, вогнутая переносица, укороченный шейный отдел позвоночника. Больные рождаются с серьезными дефектами лица — расщелиной неба и верхней губы. Подобные уродства делают больного ребенка не похожим на человека.
- Признаки поражения костей, суставов, мышц: сжатая в кулак кисть, полидактилия, сращение пальцев, деформированная стопа.
- Симптомы со стороны ЦНС: недоразвитие мозжечка, водянка головного мозга, дисгенезия мозолистого тела, грыжи различных отделов позвоночника, глухота.
- Врожденные аномалии зрительного анализатора проявляются колобомой, помутнением роговицы, микрофтальмией, врожденным помутнением хрусталика, атипичным строением сетчатки, недоразвитием зрительного нерва и его гипофункцией, отсутствием одного глаза, циклопией.
- Эндокринные нарушения — гипопитуитаризм.
- Поражение внутренних органов: пищеварения – воспаление и гетеротопия поджелудочной железы, кисты в железистой ткани, незавершенный поворот кишечника, дивертикул Меккеля, наличие добавочной селезенки; мочевыделения – увеличение почки и ее водянка, капиллярные гемангиомы и поликистоз почек, недоразвитие или дублирование мочеточника, отсутствие задней стенки уретры; сердца и сосудов — нарушение строения сердца, дефекты его перегородок, изменение нормальных объемов крупных и средних сосудов, разобщение малого и большого круга кровообращения; половых органов – крипторхизм, гипертрофия клитора, двурогая матка, недоразвитый половой член. Аномально развитая половая система — причина полной утраты репродуктивной способности, не подлежащей восстановлению.
- Младенцы нередко страдают от грыжи пупка, имеют глубокую степень умственной отсталости и входят в группу риска по развитию лейкоза.
Новорожденные с такими патологиями редко доживают до года: 95% больных умирает практически сразу после родов. Продолжительность жизни при данном синдроме в отдельных случаях может достигать пяти или десяти лет.
На основании клинических проявлений болезни и особенностей течения патологического процесса выделяют следующие формы синдрома:
- Легкая форма — ребенок рождается с особенностями внешнего вида, отличающими его от остальных детей, гипотрофией и незначительными функциональными нарушениями внутренних органов. Продолжительность жизни колеблется от нескольких месяцев до года. При отсутствии тщательного ухода и медицинского наблюдения болезнь быстро прогрессирует.
- Умеренная форма – новорожденный имеет характерный внешний вид, обезображивающий его и позволяющий поставить предварительный диагноз сразу после рождения, а также дисфункцию жизненно важных органов, нарушающую процессы жизнедеятельности. Больные дети в среднем живут 2-3 дня и погибают от развившихся осложнений. Продлить жизнь таким детям поможет только хирургическое вмешательство, устраняющее врожденные аномалии.
- Тяжелая форма — ребенок погибает внутриутробно или умирает спустя несколько часов от рождения от полиорганной недостаточности.
Диагностические мероприятия
Диагностика данной патологии, как и любого другого генетического заболевания, проводится пренатально и постнатально. Внутриутробное обнаружение подобных аномалий позволяет сопоставить возможные риски рождения больного ребенка. Методы пренатальной диагностики делятся на инвазивные и неинавазивные.
Неинвазивные методы считаются безопасными. Чтобы обнаружить генетическую аномалию, не требуется материал плода. Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока рекомендованы всем беременным женщинам. Диагностика с помощью УЗИ является достоверной на 100%.

Стандартный пренатальный скрининг включает анализ крови на биохимические маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. Если полученные данные выходят за пределы нормальных значений, врачи рекомендуют прервать беременность. Для анализа необходима венозная кровь беременной женщины, в которой имеются фрагменты генетической структуры плода.
Инвазивные методы включают:
- Биопсию ворсин хориона, позволяющую диагностировать недуг с 8 по 12 неделю беременности. Одну из плодных оболочек берут на анализ с помощью пункционной иглы. Для исследования достаточно небольшого количества материала.
- Амниоцентез — забор околоплодных вод специальной иглой через брюшину для проведения клеточного анализа. Метод актуален с 14 по 18 неделю беременности. Исследование проводится под контролем УЗИ. Клетки, содержащие ДНК плода, проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пуповинной крови плода, позволяющее определить генетические аномалии с высокой точностью. Проводится оно после 20-й недели гестации.
Несмотря на высокую точность и надежность инвазивных методов, их применяют только в крайних случаях. Внедрение в организм беременной женщины и проникновение в оболочки плода несет определенный риск. Любое неверное движение специалиста, проводящего процедуру, может привести к внутриутробной гибели плода.
Диагноз устанавливают после определения кариотипа младенца путем КФ-ПЦР. Только анализ ДНК позволяет подтвердить или опровергнуть предполагаемый диагноз. С помощью хромосомных исследований можно определить, есть ли у ребенка трисомия 13, наследственная ли у аномалии этиология или она обусловлена спонтанной внутриутробной мутацией. Чтобы оценить риски и суметь предотвратить подобные мутации, родители должны пройти подробное генетическое исследование, которое дает 100%-ный результат. Генетическая экспертиза проводится, даже если произошла внутриутробная смерть плода, с целью предотвращения синдрома при повторной беременности.
Постнатальная диагностика заключается в обследовании новорожденного неонатологом и выявлении яркой и специфичной симптоматики. Больным детям показано тщательное всестороннее обследование, включающее следующие инструментальные методики:
- Кардиографию – электрокардиографию, фонокардиографию, коронарографию, магнитокардиографию;
- УЗИ внутренних органов, расположенных в абдоминальной полости и малом тазу: поджелудочной железы, почек, мочеточников, органов репродуктивной системы;
- Нейросонографию — УЗИ головного мозга;
- Томографическое исследование мозговых структур — КТ или МРТ.
Больным детям обычно требуется консультация врачей — специалистов в области офтальмологии, отоларингологии, неврологии, детской хирургии, генетики, эндокринологии, психиатрии.
Лечение
Синдром Патау — неизлечимая хромосомная аномалия. Последствия генетических мутаций невозможно исправить. Больным детям показана симптоматическая терапия, направленная на поддержание физиологических функций организма, продление жизни и улучшение ее качества. Специалисты должны организовать хороший уход за больным ребенком, обеспечить его полноценным питанием, провести плановую иммунизацию, устранить тяжелые симптомы патологии.

Терапевтические мероприятия при синдроме Патау:
- Оперативное вмешательство по устранению расщелин на лице и удалению лишних пальцев, хирургическая коррекция работы внутренних органов,
- Консервативные меры, направленные на поддержание адекватного функционирования пораженных органов,
- Защита от инфекционных патологий,
- Общеоздоравливающие средства — прием витаминов и минералов, БАДов, иммуномодуляторов,
- Психиатрическая помощь.
Для поддержания жизнеспособности всего организма проводится коррекция работы дыхательной и пищеварительной систем, а также нормализация сердечной деятельности. Больным необходимо создать стерильные условия, чтобы избежать развития различных инфекционных заболеваний. Дети с данным синдромом требуют тщательного ухода и регулярного прохождения медицинского обследования.
Пупочные и паховые грыжи, пороки сердца и прочие аномалии лечат хирургическим путем, если состояние больного ребенка остается удовлетворительным. Оперативное вмешательство несет неоправданный риск. Хирурги, исправив внешние дефекты, могут потерять пациента из-за сбоя в работе сердечно-сосудистой системы и развития прочих серьезных осложнений.
Прогноз при данном заболевании неблагоприятный. Синдром Патау, выявленный пренатально, требует принятия важного решения о прерывании беременности. Врачи должны объяснить будущим родителям, что у них родится необычный ребенок, который никогда не сможет жить полноценной жизнью. Если родители, все обдумав, выбрали продолжение беременности, они должны помнить, что синдром Патау является смертельным расстройством. Новорожденные умирают в первые недели или месяцы жизни. Прожить полноценную и долгую жизнь могут лишь те дети, у которых врожденные аномалии выражены минимально и позволяют организму активно развиваться. Они страдают тяжелой формой идиотии, которая мешает адаптироваться к жизни в социуме, и до смерти остаются полными инвалидами, требующими заботы, опеки и особого внимания.
Профилактические мероприятия при синдроме Патау абсолютно бессмысленны, поскольку в основе патологии лежит спонтанная мутация генов. Если в роду были замечены какие-либо генетические отклонения, то перед зачатием семейной паре необходимо пройти тщательное обследование и получить консультацию у генетика.
Источник