Как выглядят дети с синдромом эдвардса

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015;
проверки требуют 16 правок.
Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21[3] и 13[4]. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%[5].
Причины заболевания[править | править код]
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома[править | править код]
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз[править | править код]
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления[править | править код]
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации[править | править код]
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
См. также[править | править код]
- Анеуплоидия
- Хромосомные болезни
- Синдром Дауна
- Синдром Патау
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Синдром Дауна
- ↑ Синдром Патау
- ↑ Genetics Home Reference. Trisomy 18 (англ.). Genetics Home Reference. Дата обращения 20 сентября 2019.
Ссылки[править | править код]
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
Сегодня мы поговорим о достаточно редком детском заболевании, сопровождающемся большим числом аномалий и нарушений в развитии. Речь пойдет о синдроме Эдвардса. Разберем его причины, формы, частоту проявления, методы диагностики и прочие важные вопросы.
Что это?
Синдром Эдвардса — болезнь, обусловленная хромосомными аномалиями, вызывающая целый список нарушений и отклонений в развитии ребенка. Ее причина — трисомия 18-й хромосомы, т. е. присутствие ее лишней копии. Этот факт и ведет к появлению осложнений генетической природы.
Риск того, что родится ребенок с синдромом Эдвардса, равен 1 случаю из 7000. К сожалению, большинство младенцев с данным отклонением умирают в первые недели жизни. Только порядка 10 % живут один год. Болезнь ведет за собой глубокую умственную отсталость, врожденные поражения внутренних и внешних органов. Самые частые из них — порок мозга, сердца, почек, маленькая голова и челюсть, расщелина губы или неба, косолапость.

Впервые сформированы и описаны симптомы заболевания были в 1960 году Д. Эдвардсом. Доктор сумел установить зависимость между проявлением нескольких признаков, обнаружил более 130 дефектов, которые сопутствуют заболеванию. Хоть симптомы синдрома Эдвардса проявляются весьма ярко, современные методы терапии против них, к сожалению, бессильны.
Причина заболевания
Если синдром Эдвардса (фото больных детей по этическим соображениям не будут размещены) был диагностирован во время беременности, то чаще всего последняя заканчивается выкидышем или мертворождением. Увы, проявление болезни у плода предотвратить сегодня нельзя.
Также в современности не выяснены четкие причины этого генетического заболевания, отчего и профилактические меры против развития его у будущих детей сформировать нельзя. Однако специалисты определили факторы риска:
- Неблагоприятные экологические условия.
- Воздействие радиации, токсических, химических веществ на родителей.
- Пристрастие к алкоголю и табаку.
- Наследственность.
- Прием определенных медикаментов.
- Инцест, кровное родство родителей.
- Возраст будущей матери. Если женщине старше 35 лет, то это считается причиной синдрома Эдвардса у ребенка, равно как и иных хромосомных заболеваний.
Формы синдрома
На тип такой аномалии в первую очередь влияет стадия развития зародыша, на которой синдром настигает эмбрион. Всего различаются три вида:
- Полный. Самый тяжелый тип, на него приходится 80 % случаев. Утроенная хромосома появляется в тот момент, когда плод был всего одной клеткой. Отсюда следует, что аномальный хромосомный набор будет передаваться при делении и всем остальным клеткам, наблюдаться в каждой из них.
- Мозаичный. Название дано из-за того, что здоровые и мутировавшие клетки смешаны, как мозаика. 10 % пораженных симптомом Эдвардса страдают именно этой формой. Признаки болезни здесь выявлены слабее, но все же мешают нормальному развитию ребенка. Избыточная хромосома появляется во время фазы, когда зародыш состоит из нескольких клеток, поэтому поражается только часть организма или отдельный орган.
- Возможная транслокация. Здесь наблюдается не только нерасхождение хромосом, но и переизбыток информации, порожденный транслокационной перестройкой. Проявляется как во время созревания гамет, так и во время развития зародыша. Отклонения здесь не ярко выражены.
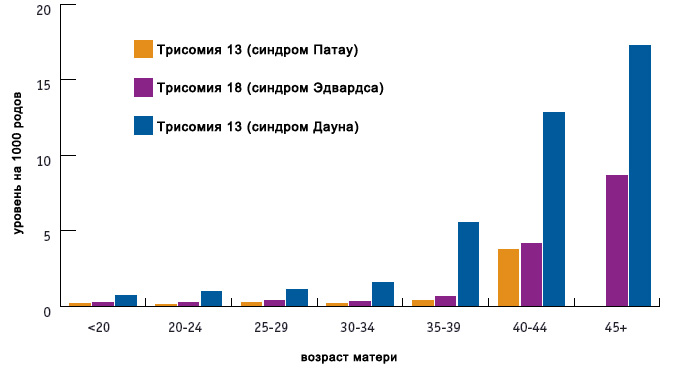
Распространенность синдрома
Риск синдрома Эдвардса невозможно выразить в точных цифрах. Нижняя граница рождения ребенка с такой аномалией — 1:10000, верхняя — 1:3300. При этом встречается он в 10 раз реже, чем синдром Дауна. Средние показатели зачатия детей с болезнью Эдвардса выше — 1:3000.
Согласно исследованиям, риск родить малыша с таким синдромом повышается при возрасте родителей более 45 лет на 0,7 %. Но он и присутствует и у 20-, 25-, 30-летних родителей. Средний возраст отца ребенка с синдромом Эдвардса — 35 лет, матери — 32,5 лет.
Аномалия также связана с полом. Доказано, что у девочек она встречается в 3 раза чаще, чем у мальчиков.
Синдром и беременность
Проявляет свои признаки синдром Эдвардса еще на стадии беременности. Последняя протекает с рядом осложнений, характерна перенашиванием — малыши рождаются примерно на 42-й неделе.
На этапе беременности заболевание плода характеризует следующее:
- Недостаточная активность эмбриона.
- Брадикардия — сниженная частота сердечных сокращений.
- Многоводие.
- Несоответствие размера плаценты размеру плода — она имеет меньший размер.
- Развитие одной пупочной артерии вместо двух, что ведет к кислородной недостаточности, асфиксии.
- Грыжа брюшной полости.
- Сплетение сосудистых образований, видимое на УЗИ (обнаруживаются у 30 % детей, пораженных синдромом).
- Недостаточный вес плода.
- Гипотрофия — хроническое расстройство функций ЖКТ.
60 % детей погибают уже в материнской утробе.
Дородовая диагностика
Синдром Эдвардса на УЗИ возможно определить только по косвенным признакам. Самый точный метод диагностики синдрома у плода сегодня — перинатальный скрининг. На его основе при возникновении тревожных подозрений доктор уже направляет женщину на инвазивное тестирование.
Скрининг, выявляющий кариотип синдрома Эдвардса, разделяется на два этапа:
- Первый проводится на 11-13 неделе беременности. Исследуются биохимические показатели — проверяется кровь матери на уровень гормонов. Результаты на этом этапе не окончательны — они могут говорить только о наличии риска. Для расчетов специалисту нужен белок А, ХГЧ, белок, вырабатываемый оболочками эмбриона и плаценты.
- Второй этап уже направлен на точный результат. Для исследований берется образец пуповинной крови или околоплодной жидкости, который затем повергается генетическому анализу.

Инвазивное тестирование
Хромосомы синдрома Эдвардса вероятнее всего определить данным методом. Однако он обязательно предполагает оперативное вмешательство и проникновение в оболочки эмбриона. Отсюда риск прерывания беременности и развития осложнений, отчего тест назначается только в крайних случаях.
На сегодня известны три типа взятия образца:
- БВХ (биопсия ворсин хориона). Основное преимущество метода — образец берется, начиная с 8-й недели беременности, что позволяет на ранних сроках определить осложнения. Для исследований нужен образец хориона (один из слоев оболочки плаценты), структура которого схожа со структурой эмбриона. Данный материал позволяет диагностировать внутриутробные инфекции, генетические и хромосомные болезни.
- Амниоцентез. Анализ проводится, начиная с 14-й недели беременности. В этом случае зондом протыкаются амниотические оболочки эмбриона, инструмент собирает образец околоплодных вод, содержащих в себе клетки будущего ребенка. Риск развития осложнений от такого исследования гораздо выше, чем в предыдущем случае.
- Кордоцентез. Срок — не ранее 20-й недели. Здесь берется образец пуповинной крови эмбриона. Сложность в том, что при взятии материала у специалиста нет права на ошибку — он должен попасть иглой точно в сосуд пуповины. На практике это происходит так: через переднюю стенку брюшины женщины вводится пункционная игла, которая собирает порядка 5 мл крови. Процедура проходит под контролем УЗИ-устройств.
Все вышеописанные методы нельзя назвать безболезненными и безопасными. Поэтому их проводят только в случаях, когда риск генетического заболевания у плода выше риска развития осложнений от взятия материала для анализа.
Родителям надо помнить, что ошибка медика при процедуре может привести к проявлению серьезных заболеваний, врожденных пороков у будущего ребенка. Нельзя исключить и риск внезапного прерывания беременности от подобного вмешательства.
Неинвазивное тестирование
Диагностика синдрома Эдвардса у плода включает в себя и неинвазивные методы. То есть без проникновения в плодные оболочки. Притом в точности такие методы не уступают инвазивным.
Одним из высокоточных анализов данного типа можно назвать кариотипирование. Это взятие образца крови матери, содержащей в себе свободные ДНК эмбриона. Специалисты извлекают их из материала, копируют, после чего проводят необходимые исследования.
Послеродовое диагностирование
Специалист может определить детей с синдромом Эдвардса и по внешним признакам. Однако для постановке окончательного диагноза проводятся следующие процедуры:
- УЗИ — исследование патологий внутренних органов, обязательно сердца.
- Томография головного мозга.
- Консультация детского хирурга.
- Обследования у специалистов — эндокринолога, невролога, отоларинголога, офтальмолога, ранее работавших с детьми, страдающими от данного заболевания.
Отклонения при синдроме
Патологии, причина которых — трисомия по 18 хромосоме, довольно серьезны. Поэтому только 10 % детей с синдромом Эдвардса доживают до года. В основном же девочки живут не более 280 суток, мальчики — не более 60.
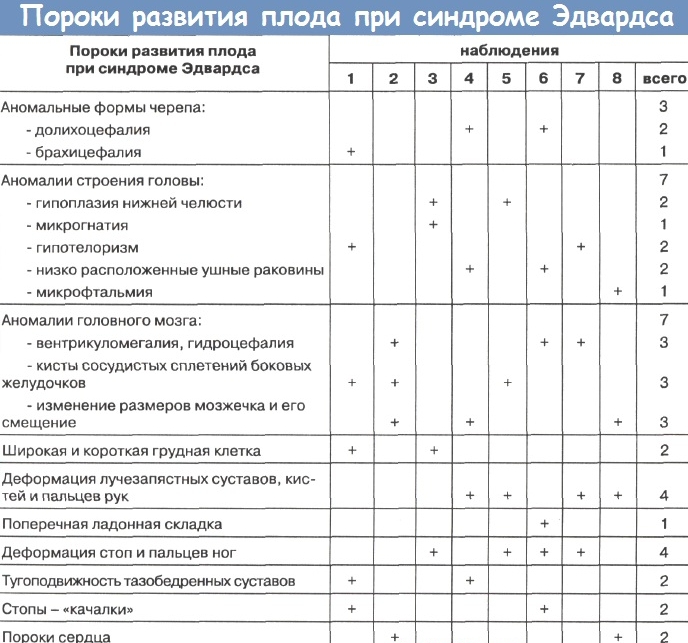
У детей наблюдаются следующие внешние отклонения:
- Вытянутый от макушки к подбородку череп.
- Микроцефалия (маленький размер головы и мозга).
- Гидроцефалия (скопление жидкости в черепной коробке).
- Узкий лоб при широком затылке.
- Аномально низкое расположение ушей. Может отсутствовать мочка или козелок.
- Укороченная верхняя губа, делающая рот треугольным.
- Высокое небо, часто со щелью.
- Деформированные челюстные кости — нижняя челюсть аномально маленькая, узкая и неразвитая.
- Укроченная шея.
- Аномально узкие и короткие глазные щели.
- Отсутствие части глазной оболочки, катаракта, колобома.
- Нарушение функций суставов.
- Недоразвитые, малоподвижные стопы.
- Из-за аномального строения пальцев могут формироваться ластообразные конечности.
- Порок сердца.
- Ненормально расширенная грудная клетка.
- Нарушенная работа эндокринной системы, в частности, надпочечников и щитовидной железы.
- Необычное расположение кишечника.
- Неправильная форма почек.
- Удвоение мочеточника.
- У мальчиков — крипторхизм, у девочек — гипертрофированный клитор.
Отклонения психического характера обычно следующие:
- Недостаточно развитый головной мозг.
- Осложненная степень олигофрении.
- Судорожный синдром.

Прогноз для больных синдромом Эдвардса
К сожалению, на сегодня прогнозы неутешительны — порядка 95 % детей с данной болезнью не доживают до 12 месяцев. При этом тяжесть ее формы не зависит от соотношения больных и здоровых клеток. У выживших детей отмечаются отклонения физической природы, тяжелая степень олигофрении. Жизнедеятельность такого ребенка нуждается во всестороннем контроле и поддержке.
Нередко дети с синдромом Эдвардса (фото не представлены в статье из этических соображений) начинают различать эмоции окружающих, улыбаться. Но их общение, умственное развитие при этом ограничено. Ребенок со временем может научиться сам поднимать голову, кушать.
Возможности терапии
Сегодня такое генетическое заболевание неизлечимо. Ребенку назначается только поддерживающая его состояние терапия. Жизнь пациента связана со многими аномалиями и осложнениями:
- Мышечная атрофия.
- Косоглазие.
- Сколиоз.
- Сбои в работе сердечно-сосудистой системы.
- Атония кишечника.
- Низкий тонус стенок брюшины.
- Отит.
- Пневмония.
- Конъюнктивит.
- Синусит.
- Заболевания мочеполовой системы.
- Большая вероятность развития рака почки.
Заключение
Подводя итог, хочется отметить, что синдром Эдвардса не передается по наследству. Больные в большинстве случаев не доживают до репродуктивного возраста. При этом они не способны к продолжению рода — для заболевания характерна недоразвитость половой системы. Что касается родителей ребенка с синдромом Эдвардса, то шанс постановки такого же диагноза при следующей беременности равен 0,01 %. Надо сказать, что и само заболевание проявляет себя весьма редко — диагностируется только у 1% новорожденных. Нет и особых причин для его возникновения — в большей части случаев родители совершенно здоровы.
Источник
Причины
23 пары хромосом, унаследованных от родителей, в норме содержит каждая клетка организма человека. Когда соединяются сперматозоид и яйцеклетка, образуя эмбрион, объединяются их хромосомы. Ребёнок получает 23 хромосомы из сперматозоида отца и 23 от яйца матери — всего 46.
Иногда яйцеклетка или сперматозоид имеют неверное число хромосом. Поскольку клетки отца и матери соединяются, ребёнку передаётся это искажение.
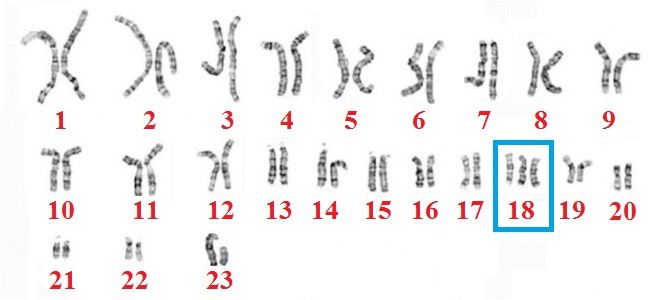
«Трисомия» значит, что ребёнок имеет дополнительную хромосому во всех или некоторых клетках. При трисомии 18 у малыша есть три хромосомы 18. Это приводит к ненормальному развитию многих органов у крохи.
Как правило, трисомия 18 вызвана содержанием лишней хромосомы 18 в каждой клетке. Около 5% пострадавших людей имеют дополнительную хромосому 18 в отдельных, а не во всех клетках. Это мозаичная трисомия. Она может быть очень тяжёлой или едва заметной, в зависимости от числа клеток, имеющих лишнюю хромосому.
В редких случаях нет дополнительной хромосомы; часть длинной ветви хромосомы 18 соединяется с другой хромосомой во время образования сперматозоидов и яйцеклетки или в начале развития эмбриона. В этом случае индивидуум имеет 2 хромосомы 18 и ещё добавочный материал из хромосомы 18, который прикреплён к другой хромосоме. Это явление получило название транслокация.
Дополнительный генетический материал вызывает аномалии развития так же, как наличие всей дополнительной хромосомы. Признаки и симптомы этой формы трисомии зависят от количества хромосомного материала, который был перенесён в другую хромосому.
О механизме наследования
Большинство случаев синдрома не наследуются. Он возникает из-за случайных событий во время образования яйцеклетки и спермы. В результате ошибки в делении возникает репродуктивная клетка с неправильным числом хромосом. Например, у сперматозоида или яйцеклетки может появиться добавочная копия хромосомы 18. Когда одна из таких нетипичных репродуктивных клеток вносит вклад в генетический состав ребёнка, у него во всех клетках будет дополнительная хромосома 18.
Также не наследуется мозаичная трисомия 18. Она обусловлена случайными событиями при делении клеток в начале эмбрионального развития. В итоге отдельные клетки содержат две копии хромосомы 18, а другие — три копии этой хромосомы.
Может быть унаследована транслокационная трисомия. Незатронутый человек несёт перегруппировку генетического материала между хромосомой 18 и другой хромосомой. Хотя у него нет признаков трисомии 18, человек, у которого есть этот тип транслокации, подвергаются повышенному риску иметь детей с этим генетическим расстройством.
Эпидемиология
Синдром Эдвардса является второй по частоте аутосомной трисомией среди живых детей после синдрома Дауна.
Трисомия 18 встречается, в среднем, у 1 из каждых 5000 детей. Девочки страдают гораздо чаще, чем мальчики. У любой женщины может родиться ребёнок с трисомией 18, но риск возрастает с увеличением возраста матери.
Признаки синдрома Эдвардса
У детей, рождённых с трисомией 18, могут быть некоторые или все из этих характеристик:
- пороки развития почек;
- структурные дефекты сердца при рождении (дефект межжелудочковой и предсердной перегородок, открытый артериальный проток);
- кишечник, выступающий вне тела (омфалоцеле);
- атрезия пищевода (непроходимость пищевода);
- умственная отсталость;
- отставание в развитии;
- дефицит роста;
- трудности с кормлением;
- трудности с дыханием;
- артрогрипоз (тугоподвижность суставов).
Некоторые физические пороки развития, ассоциированные с синдромом Эдвардса:
- небольшая голова (микроцефалия);
- низко расположенные, неправильно сформированные уши;
- аномально маленькая челюсть (микрогнатия);
- расщелина губы / волчья пасть;
- перевёрнутый нос;
- узкие, широко посаженные глаза (глазной гипертелоризм);
- опускание верхних век (птоз);
- короткая грудная кость;
- сжатые руки;
- недоразвитые большие пальцы и / или ногти;
- сращение второго и третьего пальцев ноги;
- косолапость;
- у мальчиков неопущенние яичек.
В утробе наиболее распространённой характеристикой являются аномалии сердца и ЦНС. Наиболее частая внутричерепная патология — наличие кист хориоидного сплетения, которые являются карманами жидкости в мозге. Иногда проявляется избыток амниотической жидкости.
Диагностика
У новорождённых хромосомный анализ может быть проведён для определения точной причины врождённых дефектов.
Возможны пренатальные исследования для трисомии 18:
- Измерение уровня альфафетопротеина. Тест проводится между 15 и 17 неделями беременности. Положительный результат исследования не означает, что у ребёнка будет трисомия 18 или какая-либо хромосомная аномалия. Фактически, только около у 11% тех женщин с положительным результатом на трисомию 18 в этом тесте, действительно будет пострадавший плод.
- Ультразвук — ещё один широко используемый скрининг-тест. Как и выше описанное исследование, простое УЗИ не может быть использовано для установления диагноза трисомии 18. Более подробное исследование при помощи ультразвуковых волн может быть выполнено для поиска характерных признаков аномалии, но этот метод не может подтвердить наличие синдрома.
- Анализ эмбрионального хромосомного материала, полученного при амниоцентезе или биопсии ворсин хорион, необходим, чтобы доказать, что имеется дополнительная копия хромосомы 18. Амниоцентез обычно проводится на 15 — 18 неделе беременности и является наиболее часто используемым тестом для пренатальной диагностики трисомии 18. Во время этой процедуры через брюшную стенку вводят тонкую иглу и берут небольшой образец амниотической жидкости. Биопсия ворсин хориона является ещё одним типом исследования, который позволяет учить генетический материал плода. Тест выполняется через 10 — 12 недель после последнего менструального цикла и, следовательно, имеет преимущество, позволяющее установить более ранний диагноз. Эта процедура включает в себя сбор образца ворсин хориона из плаценты с помощью прокола брюшной стенки, либо с использованием катетера через влагалище.
Лечение
В настоящее время медицинская наука не нашла лекарство от синдрома Эдварда. У младенцев с синдромом обычно присутствуют серьёзные физические нарушения, и врачи сталкиваются с трудностью выбора в отношении их лечения. Хирургия может помочь в лечении некоторых из проблем, связанных с синдромом. Сегодня лечение в основном состоит из паллиативной помощи (поддержание и улучшение качества жизни пострадавших пациентов).
Приблизительно 5 – 10 % детей с синдромом Эдварда выживают после первого года жизни благодаря лечению различных хронических патологий, связанных с синдромом. Проблемы, ассоциированные с аномалиями нервной системы и мышечным тонусом, влияют на развитие моторных навыков у младенца, что может привести к сколиозу и косоглазию. Применение методов хирургического вмешательства ограничено из-за присутствующих заболеваний сердца.
У младенцев с синдромом Эдварда возможен запор, возникший из-за плохого тонуса брюшной мышцы. Результатом этого могут быть дискомфорт, раздражительность и проблемы с питанием. Специальные молочные смеси, препараты от газов, слабительные средства, размягчители стула, а также суппозитории — это потенциальные методы лечения, которые может рекомендовать врач.
Клизмы не рекомендуются, так как они могут выводить электролиты и изменять состав жидкости в организме.
У пострадавших детей наблюдаются серьёзные задержки развития, хотя с ранним вмешательством посредством программ терапии и специального образования они могут достичь определённых этапов развития. У таких пациентов есть повышенный риск возникновения «опухоли Вильмса» — это форма рака почки, которая по большей части поражает детей. Рекомендуется регулярно выполнять УЗИ органов брюшной полости.
Заключение
Средняя длительность жизни для половины детей, родившихся с этим синдромом, составляет менее двух месяцев; приблизительно 90 – 95 % этих детей умирают до первого дня рождения. У 5 — 10 % пациентов, переживших свой первый год, есть серьёзные нарушения в развитии.
Дети, дожившие до года, нуждаются в поддержке при ходьбе, и их способность учиться ограничена. Вербальные коммуникативные способности также ограничены, хотя они способны реагировать на утешение, и могут научиться улыбаться, распознавать родителей и других людей, взаимодействовать с ними.
Источник