Эктопия миндалин мозжечка код мкб 10

Рубрика МКБ-10: Q07.0
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q00-Q07 Врожденные аномалии пороки развития нервной системы / Q07 Другие врожденные аномалии пороки развития нервной системы
Определение и общие сведения[править]
Аномалия (мальформация) Арнольда-Киари — одно из многочисленных врождённых заболеваний краниовертебральной области.
В 1891 г. Киари описал три различных вида мальформаций, сочетаемых с гидроцефалией.
Эпидемиология
Среди известных трех типов аномалии Арнольда-Киари у взрослых особый интерес вызывает I тип. Частота встречаемости данной аномалии I типа составляет 3,8-8,2 на 1000 населения. Соотношение мужчин и женщин, по данным ряда авторов, приблизительно одинаковое, однако несколько преобладают женщины — 59%. В связи с современными возможностями прижизненной нейровизуализации стали часто выявлять такую аномалию. По некоторым данным, мальформацию Арнольда-Киари обнаруживают среди больных с различной патологией нервной системы при магнитно-резонансной томографии у 26% пациентов.
Классификация
1. Аномалия Арнольда-Киари I (АК I)
Аномалия Арнольда-Киари I представляет тонзиллярную эктопию, при которой деформированная миндалина мозжечка смещена ниже уровня большого затылочного отверстия в верхнюю часть цервикального канала.
При смещении миндалин мозжечка на расстояние 5 мм и более возникают начальные клинические признаки. У детей в возрасте 5-15 лет миндалины мозжечка расположены более низко, чем у взрослых и детей до 5 лет. Асимптоматичную эктопию миндалин мозжечка на расстояние 6 мм ниже уровня большого затылочного отверстия у детей 5-15 лет не должны считать патологической. Аномалия Арнольда-Киари I (АК I) в 20-25% случаев связана с сирингогидромиелией и умеренной гидроцефалией. Часто сочетается с аномалией краниовертебральной области: базиллярной импрессией, незавершенной оссификацией и сращением позвонка С1 с затылочной костью, синдромом Клиппеля-Фейля.
2. Аномалия Арнольда-Киари II
Комплекс аномалии ствола мозга, спинного мозга, костей черепа и позвоночника. Изменения головного мозга и черепа определяют в виде низкого расположения стока синусов и поперечных синусов. Следствие этого — маленькая и мелкая задняя черепная ямка, расширенное большое затылочное отверстие, тенториальная гипоплазия с широкой вырезкой, червь и мозжечок могут пролабировать вверх через тенториальную вырезку. Дизгенезия ствола мозга ведет к смещению вниз продолговатого мозга и мозжечка. Происходят изгиб продолговатого мозга, выпячивание покрышки, переднемедиальный рост мозжечка по бокам ствола мозга.
Аномалии развития задней черепной ямки приводят к нарушению ликворциркуляции, что служит причиной окклюзионной гидроцефалии. Часто увеличен III желудочек с деформированным передним карманом. Удлинен IV желудочек, он имеет небольшие размеры и смещен каудально. Могут определяться стеноз или окклюзия водопровода, а также изолированный IV желудочек, который в большинстве случаев сочетается с сирингогидромиелией.
АК II часто сочетается с пороками развития, локализуемыми супратенториально. К ним относят аномалии мозолистого тела, мальформации кортикального развития, увеличение размера хвостатого ядра, фенестрацию серпа большого мозга.
Спинальный дисрафизм и миеломенингоцеле присутствуют почти всегда. Наиболее частая патология — миеломенингоцеле. В 75% случаев поражен пояснично-крестцовый отдел позвоночника. В спинном мозге могут определять сирингогидромиелию. Поражение спинного мозга может быть обусловлено сдавлением липомой, диастематомиелией.
3. Аномалию Арнольда-Киари III встречают очень редко. Она представлена аномалией АК II с низким затылочным и высоким шейным энцефалоцеле.
Этиология и патогенез[править]
Большинство авторов считают, что основную роль в развитии аномалии играет патология внутриутробного развития костных и мягкотканных структур задней черепной ямки. В клинике нередко встречают семейные случаи аномалии Арнольда-Киари I типа, и при анализе их наиболее вероятен аутосомно-доминантный или аутосомно-рецессивный тип наследования. Существует гипотеза о генетической природе заболевания, связанной, возможно, с экспрессией Pax-1 и Pax-9 генов, определяющих развитие аксиальных образований.
Однако есть публикации, в которых всё более убедительно доказывают роль родовой травмы в развитии данной патологии. В результате проведённых морфометрических исследований костных и невральных образований задней черепной ямки у больных с аномалией Арнольда-Киари I типа отмечали наличие диспропорции между длиной блюменбахова ската и ствола мозга. Эту диспропорцию авторы объясняют перенесённой родовой травмой. Первоначально происходит повреждение клиновидно-решетчатого и клиновидно-затылочного синхондрозов. Костные отломки при постоянной их подвижности из-за пульсации мозга не могут срастись между собой. Отсутствие прочной фиксации костных отломков делает условия заживления неблагоприятными. Формирование и обызвествление костных структур идут медленно и неправильно. Это ведёт к укорочению и деформации кливуса и, как следствие, нарушает в постнатальном периоде формирование задней черепной ямки.
Очевидно хронические грыжи заднего мозга при врождённых пороках относятся к дизэмбриогенезиям, т.е. генетически детерминированы и имеют наследственно-обусловленную природу. В основе мальформации Арнольда-Киари лежит диспропорция между объёмом невральных образований и вместимостью задней черепной ямки, запрограммированной дефектным развитием парааксиальной мезодермы.
Одна из наиболее тяжёлых клинических ситуаций — сочетание данной аномалии и сирингомиелии. Большинство авторов считают образование сирингомиелических кист осложнением аномалии Арнольда-Киари.
Некоторые исследователи указывают, что сирингомиелия сочетается с данной аномалией в 48-76% наблюдений. К осложнениям аномалии относят также и гидроцефалию, возникающую вследствие длительно нарастающих нарушений ликвородинамики.
В зависимости от причины нарушений ликвороциркуляции при аномалии I типа предложена их классификация с выделением изолированной формы мальформации Арнольда-Киари, мальформации в сочетании с сирингомиелией, мальформации с гидроцефалией и мальформации с патологическим объёмом спинномозговой жидкости в области задней черепной ямки.
Клинические проявления[править]
Клинические проявления аномалии возникают не внезапно, постепенно нарастая в выраженности. Часто первые симптомы появляются уже в детском или молодом возрасте, реже заболевание дебютирует в зрелом возрасте.
Как правило, пациенты с краниовертебральными аномалиями, в т.ч. с аномалией Арнольда-Киари, имеют дизрафический статус: короткая шея, шейные рёбра, низкая граница роста волос в шейно-затылочной области, асимметрия лица и черепа, готическое нёбо, приращение мочек уха, кифосколиоз позвоночника, реберный горб, неравномерное стояние лопаток, воронкообразная грудь, плоскостопие и др.
Больные жалуются на боли в шейно-затылочной области, головокружение, шаткость при ходьбе, нарушения глотания и фонации, онемение и слабость в конечностях, иногда у них возникают синкопальные пароксизмы.
Клиническая картина аномалии Арнольда-Киари полиморфна. Она включает сочетание мозжечковых, спинальных, бульбарных расстройств, сопровождающихся симптомами внутричерепной гипертензии.
До настоящего времени отсутствует единая синдромальная классификация клинических проявлений аномалии Арнольда-Киари. М.Д. Благодатским и соавт. была предложена классификация синдромов: гипертензионногидроцефальный, пирамидный, бульбарный, синдром интрамедуллярного поражения.
Пирамидный синдром может быть представлен тетра-, пара- или гемипарезом.
Бульбарный синдром характеризуется одноили двусторонним поражением каудальной группы черепных нервов. У больных развиваются нарушения глотания и фонации, снижение или отсутствие глоточного рефлекса, атрофия мышц половины языка.
Симптомы интрамедуллярного поражения свидетельствуют о развитии гидро- и сирингомиелии. Авторы почти во всех случаях сирингомиелии обнаруживали у больных кифосколиоз, обусловленный поражением дорсомедиальных и вентромедиальных ядер с развитием пареза аксиальной мускулатуры и трофических нарушений.
Реже в клинической картине наблюдают проявления поражения V, VI, VII нервов: невралгию тройничного нерва, глазодвигательные нарушения, асимметрию лицевой мускулатуры. Причиной дисфункции черепных нервов может быть в ряде случаев нейроваскулярная компрессия при сочетании мальформации Арнольда-Киари с аномалией сосудов головного мозга, и в том числе петлеобразованием задней нижней мозжечковой артерии. Это вызывает такие проявления дисфункции черепных нервов, как тригеминальная невралгия, гемифациальный спазм, языкоглоточные боли. Компрессионное воздействие задней нижней мозжечковой артерии на продолговатый мозг при аномалии Арнольда-Киари может быть причиной артериальной гипертензии вследствие постоянного раздражения пульсирующей артерией сосудодвигательного центра в области нижнего отдела ромбовидной ямки.
Часто при аномалии определяют корешковые нарушения. Характерный признак аномалии Арнольда-Киари I типа — гипотрофия трапециевидных мышц, обусловленная нарушением проводимости по добавочным нервам, подтверждаемого данными электромиографии.
Нередко отмечают сочетание аномалии Арнольда-Киари и аномалии краниовертебральной области (платибазия и базилярная импрессия). Наиболее грубое поражение продолговатого мозга и верхнешейных сегментов спинного мозга наблюдают при базилярной импрессии, когда сдавление вентральной поверхности мозга вызывает клинические проявления тяжёлого поперечного поражения.
Синдром Арнольда-Киари: Диагностика[править]
МРТ
1. Аномалия Арнольда-Киари I
При проведении МРТ на срединном сагиттальном срезе, лучше на Т1-ВИ, определяют удлиненные миндалины мозжечка, расположенные ниже плоскости большого затылочного отверстия. Пролапс должен быть не менее 4 мм. Большая затылочная цистерна мала или отсутствует. Часто определяют также смещение ствола мозга кпереди и сглаженность моста. У пациентов с атипичными клиническими проявлениями и недостоверными данными, полученными при стандартном исследовании, нарушения ликворциркуляции могут обнаружить с помощью фазоконтрастных методик. Нарушения ликворциркуляции часто локализованы на уровне цистерны моста и большого затылочного отверстия.
2. Аномалия Арнольда-Киари II
На срединном сагиттальном срезе хорошо видна небольшая задняя черепная ямка треугольной или конической формы. Она плотно выполнена мозжечком и структурами ствола мозга. Мост уплощен, прилежит к скату, зачастую истончен. Вследствие дисплазии намета мозжечка может произойти восходящее вклинение мозжечка. Переднезадний размер IV желудочка уменьшен, может отмечаться смещение его вниз. Продолговатый мозг и мозжечок через расширенное большое затылочное отверстие смещены в шейную дуральную воронку. Иногда отмечают изгиб продолговатого мозга в сагиттальной плоскости. В 50% случаев присутствует сирингогидромиелия.
Супратенториально одна из наиболее часто встречаемых сочетанных аномалий — дисгенезия мозолистого тела. Супратенториальные отделы желудочковой системы расширены. Объем промежуточных ядер может быть увеличен. Расширение затылочных рогов сохраняется и после проведения шунтирующих операций.
КТ
Определяют расширение желудочков мозга различной степени выраженности. На своем привычном месте не визуализируют IV желудочек из-за его смещения книзу. Протрузию миндалин мозжечка в большое затылочное отверстие и степень смещения зачастую трудно определить. Расширение большого затылочного отверстия и позвоночного канала. Можно обнаружить гипоплазию серпа и намета мозжечка. Средний мозг удлинен каудально и инвагинирует в мозжечок. Верхние отделы мозжечка могут смещаться супратенториально через гипоплазированную вырезку намета мозжечка.
УЗИ
Аномалию АК I можно заподозрить при исследовании головного мозга плода в сагиттальной плоскости, когда большая цистерна мозга резко уменьшена или ее не визуализируют. Форма мозжечка при этом не меняется.
Для аномалии АК II при УЗИ характерно отсутствие изображения большой цистерны в горизонтальной и сагиттальной плоскости, мозжечок деформирован и смещен каудально. Критерии спинномозговой грыжи плода при УЗИ-диагностике — нарушение целостности позвонков, отсутствие их задних дуг, кожи и мышц над дефектом. В сагиттальной плоскости возможна оценка изгибов позвоночника.
Алгоритм исследования
Пренатальная диагностика аномалии АК I не имеет четких эхографических характеристик и поэтому крайне субъективна.
Дифференциальный диагноз[править]
Диагностика аномалии АК I в большинстве случаев не вызывает проблем. Причиной вклинения миндалин мозжечка в большое затылочное отверстие могут быть объемный процесс в задней черепной ямке, укорочение свободного края намета мозжечка с пролабированием в заднюю черепную ямку базальных отделов гемисфер большого мозга.
Аномалия АК II всегда связана с некоторыми формами спинального дисморфизма, а также менингоцеле или миеломенингоцеле и гидроцефалией. Спинномозговую грыжу при АК II необходимо дифференцировать с крестцово-копчиковой тератомой I типа (опухоль расположена снаружи от позвоночника и не имеет пресакрального компонента). В отличие от спинномозговой грыжи при тератомах позвоночник не вовлечен в патологический процесс, при проведении цветного допплеровского картирования может быть обнаружен внутриопухолевый кровоток.
Синдром Арнольда-Киари: Лечение[править]
Если пациент не высказывает существенных жалоб, кроме интенсивных болезненных ощущений, то ему назначается медикаментозное лечение с различными комбинациями нестероидных противовоспалительных средств, ноотропов и препаратов миорелаксирующего действия.
Наиболее распространенным хирургическим вмешательством при синдроме Арнольда-Киари является субокципитальная краниэктомия – расширение большого затылочного отверстия методом распиливания элемента затылочной кости с удалением дужки шейного позвонка. В результате операции снижается непосредственное давление на ствол головного мозга и стабилизируется циркуляция ликвора.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Лучевая диагностика и терапия заболеваний головы и шеи [Электронный ресурс] / Трофимова Т.Н. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970425695.html
Краниовертебральная патология [Электронный ресурс] / Под ред. Д.К. Богородинского, А.А. Скоромца — М. : ГЭОТАР-Медиа, 2008.
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Синдром Арнольда-Киари.
Названия
Название: Аномалия Киари.
Аномалия Киари
Синонимы диагноза
Синдром Арнольда-Киари.
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
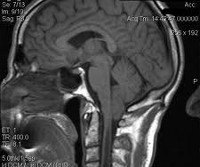
Аномалия Киари
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Симптомы
Боль в шее. Боль в шейном отделе позвоночника. Кашель. Рвота. Слабость мышц (парез).
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник