Импринтинг синдром прадера вилли и энгельмана

Синдром Пра́дера — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин[2].
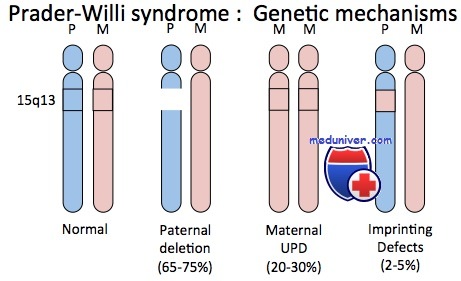
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat, когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13[3].
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер (A. Prader), Х. Вилли (H. Willi) и А. Лабхарт (A. Labhart)[4].
Особенности[править | править код]
Для синдрома Прадера — Вилли характерны:
- до рождения — низкая подвижность плода, часто — неправильное положение плода;
- дисплазия тазобедренных суставов;
- ожирение; склонность к перееданию (чаще проявляется к двум годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм) и в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.
Диагностика[править | править код]
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом).
Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Лечение[править | править код]
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Показан приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов).
Гипогонадизм обычно проявляется в микропении (микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков (крипторхизм); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск[править | править код]
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
Перспективы развития[править | править код]
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития.
Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание.
Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина. Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса[6]
См. также[править | править код]
- Синдром Мёбиуса
- Синдром Ангельмана
Примечания[править | править код]
Ссылки[править | править код]
- Л. З. Казанцева, П. В. Новиков, А. Н. Семячкина, Е. А. Николаева, М. Б. Курбатов, Э. В. Добрынина. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении. Московский НИИ педиатрии и детской хирургии Минздрава РФ / Рос. вестник перинатологии и педиатрии. — 1999. — № 4. — С.40-44.
- Сайт «Биология человека»
- Однородительская дисомия на сайте «Биология человека»
- О синдроме на сайте «Эндокринология» (EURODOCTOR.ru — Элитное лечение в Европе)[неавторитетный источник?]
- Форум родителей детей с этим диагнозом
- Мэтт Ридли. Геном: автобиография вида: В 23 главах. Глава из книги (Matt Ridley. Genome: The Autobiography of a Species in 23 Chapters)
- Подборка статей по теме, общение родителей (форум)
- Польза и вред от гормонов роста (реферат)[неавторитетный источник?]
- Кармен Сапиенца. Геномный импринтинг // Scientific American. Издание на русском языке. — 1990. — № 12. — С.14-20.
Источник
Синдромы Прадера-Вилли и Ангельмана. ХарактеристикаВозможно, наиболее полно изученные примеры роли геномного импринтинга при болезнях человека — синдромы Прадера-Вилли и Ангельмана. Синдром Прадера-Вилли — сравнительно частый дисморфический синдром, характеризующийся ожирением, чрезмерным и беспорядочным аппетитом, небольшими кистями и стопами, низким ростом, гипогонадизмом и умственной отсталостью. Приблизительно в 70% случаев синдрома наблюдают цитогенетическую делецию, затрагивающую проксимальный отдел длинного плеча хромосомы 15 (15q11-q13), причем только в хромосоме, унаследованной от отца больного. Таким образом, геном таких пациентов имеет генетическую информацию в области 15q11-q13, происходящую только от матерей. И наоборот, примерно у 70% пациентов с редким синдромом Ангельмана, характеризующегося необычным лицом, низким ростом, выраженным интеллектуальным отставанием, спастикой и судорогами, отмечают делецию приблизительно той же хромосомной области, но теперь в хромосоме, унаследованной от матери; т.е. пациенты с синдромом Ангельмана имеют генетическую информацию в регионе 15q11-q13, происходящую только от отцов. Эта необычная ситуация удивительным образом доказывает, что родительское происхождение генетического материала в описанных случаях (в хромосоме 15) имеет выраженное влияние на клиническое проявление дефекта.
Приблизительно 30% пациентов с синдромом Прадера-Вилли не имеют цитогенетически обнаруживаемых делеций; но у них выявлены две цитогенетически нормальные хромосомы 15, обе унаследованные от матери. Ситуация иллюстрирует однородительскую дисомию — наличие дисомной линии клеток, содержащих две хромосомы или их части, унаследованные от одного родителя. Если оба экземпляра представлены идентичной хромосомой, состояние называют изодисомией; если присутствуют разные гомологи от одного родителя — гетеродисомией. Приблизительно 3-5% пациентов с синдромом Ангельмана также имеют однородительскую дисомию, только с двумя неповрежденными хромосомами 15 отцовского происхождения. Эти пациенты служат дополнительным подтверждением того, что синдромы Прадера-Вилли и Ангельмана — результат потери соответственно отцовского или материнского вклада генов участка 15q11-q13. Кроме хромосомной делеции и однородительской дисомии, несколько пациентов с синдромами Прадера-Вилли и Ангельмана, вероятно, имеют дефект в самом центре импринтинга. В результате не происходит переключения от женского к мужскому импринтингу в сперматогенезе или от мужского к женскому в овогенезе.
Оплодотворение сперматозоидом, несущим аномально персистирующий женский импринтинг, приведет к рождению ребенка с синдромом Прадера-Вилли; оплодотворение яйцеклетки, имеющей несвойственный ей мужской импринтинг, закончится рождением ребенка с синдромом Ангельмана. Наконец, мутации в материнской копии одного гена — убиквитин-протеин лигазы Е6-АР, как оказалось, вызывают синдром Ангельмана. Ген убиквитин-протеин лигазы Е6-АР расположен в области 15q11-q13 и в норме импринтирован, экспрессируется только материнский аллель в центральной нервной системе (ЦНС). Полагают, что крупные материнские делеции области 15q11-q13 и отцовские однородительские дисомии хромосомы 15, наблюдаемые при синдроме Ангельмана, служат причиной заболевания, так как приводят к утрате материнской копии критически важного импринтированого гена. Мутации для одного импринтированного гена при синдроме Прадера-Вилли пока еще не обнаружены. — Вернуться в содержание раздела «генетика» на нашем сайте Оглавление темы «Аномалии хромосом»:
|
Источник
Синдром Прадера-Вилли (PWS) — это редкое генетическое заболевание, вызванное нарушением хромосомы № 15, которое дети наследуют от своего отца. В дополнение к легкой умственной отсталости и маленькому росту, неудовлетворительный голод, который приводит к хроническому перееданию и ожирению, является типичным симптомом. Поскольку это расстройство невозможно излечить, терапия направлена на облегчение симптомов и на облегчение жизни пациентов.
Что такое синдром Прадера-Вилли?
Синдром Прадера-Вилли, также известный как синдром PWS, является редким генетическим заболеванием. Это заболевание характеризуется недостаточной функцией гипоталамуса (промежуточные участки) и оказывает негативное влияние на правильное развитие нерва. Это затрагивает приблизительно 0,003 — 0,01% населения, и девочки и мальчики затронуты одинаково . По сообщениям, в мире в настоящее время около 400 000 пациентов, которые сталкиваются с этой болезнью
Редкое заболевание было впервые описано в 1956 году швейцарскими врачами Андреа Прадером, Генрихом Вилли и Алексисом Лабхартом , согласно которому синдром также назван сегодня. Двадцать пять лет спустя ген, ответственный за заболевание, был локализован, что значительно упростило диагностику. В настоящее время это заболевание можно обнаружить с помощью генетического тестирования.
Причины PWS
Синдром Прадера-Вилли является первым описанным заболеванием, вызванным генетическим импринтингом. Это связано с потерей экспрессии генов в области 15-й хромосомы (15q11-13), которую плод наследует от своего отца. Это может быть связано с:
- Микроделеция — критическая часть родительской 15-й хромосомы отсутствует (70% случаев)
- Дефект у родителей — обе хромосомы № 15 были унаследованы ребенком (25% случаев)
- Мутации PWS / AS на наследуемой от отца хромосоме (5% случаев)
- Транслокация части 15-й хромосомы (описаны минимальные случаи)
Вероятность того, что генетическое заболевание также возникает у родного брата больного пациента, зависит, прежде всего, от конкретной причины заболевания . Если область 15-й хромосомы полностью отсутствует или монопарентная дисомия, риск относительно невелик. Тем не менее, в случае хромосомной транслокации есть вероятность 25%, что это заболевание затронет другого ребенка, а при мутации PWS / AS — даже 50%.
Каковы симптомы синдрома Прадера-Вилли?
Что касается типичных симптомов, это генетическое расстройство у новорожденных проявляется неразвитым рефлексом всасывания и мышечной слабостью (гипотония). Другие симптомы включают косоглазие, неспособность преуспеть, усталость, апатию или слабый плач. В детстве пациенты маленького роста, и их лица приобретают определенные характеристики, такие как миндалевидные глаза, удлиненные головы или провисающие углы.
Синдром Прадера-Вилли является наиболее распространенной генетической причиной ожирения . Отсутствие аппетита и низкое потребление пищи скоро сменится ненасытным голодом , который никогда не исчезнет полностью. Причиной этой проблемы является высокий уровень грелина, гормона, ответственного за аппетит, который стимулирует чувство голода. Неконтролируемое желание пищи впоследствии приводит к перееданию и ожирению .
ДРУГИЕ ТИПИЧНЫЕ СИМПТОМЫ:
- Задержка двигательного и речевого развития
- Маленькие руки и ноги
- Легкая умственная отсталость
- Нарушения обучения
- Гипогонадизм — снижение производства гонадотропина
- Снижение производства половых гормонов
- Позднее наступление половой зрелости и малых половых органов
- Отсутствие вторичных половых признаков
- Сколиоз
Пациенты с синдромом Прадера-Вилли также характеризуются поведенческими расстройствами . Они могут проявлять упрямство, агрессию или суровое вранье, а также неконтролируемые приступы гнева, которые обычно связаны с лишением пищи, также распространены. ОКР (обсессивно-компульсивное расстройство) также типично . У пациентов возникают проблемы с повторяющимися мыслями или накоплением предметов, и даже незначительное самоповреждение не редкость.
Возможные осложнения
Гипогонадизм и ожирение могут вызвать серьезные осложнения у пациентов с синдромом Прадера-Вилли . Примеры включают в себя:
- Сахарный диабет 2 типа
- Сердечно-сосудистые заболевания
- Инфаркт миокарда
- бесплодие
Диагностика
Врач обычно подозревает, что ребенок страдает от синдрома Прадера-Вилли в соответствии с типичными симптомами, которые сопровождают болезнь. Подобно синдрому Ангелмана (вызванному повреждением 15-й хромосомы от матери), диагноз синдрома Прадера-Вилли можно поставить , проанализировав ДНК крови пациента.
Это генетическое заболевание также можно обнаружить во время беременности, а именно при генетическом исследовании околоплодных вод. Тем не менее, это в первую очередь используется для выявления более частых диагнозов, таких как синдром Дауна или Эдвардса , и, следовательно, PWS обычно более случайный.
Лечение синдрома Прадера-Вилли
Поскольку синдром Прадера-Вилли является результатом генетического расстройства, его невозможно вылечить . Тем не менее, существует ряд способов облегчить типичные симптомы и сделать жизнь пациентов намного проще. Также важным в этом случае является ранняя диагностика, которая поможет родителям хорошо подготовиться к уходу за ребенком с синдромом Прадера-Вилли. Таким образом, они могут заранее удовлетворить его специфические потребности и ознакомиться с важными службами вмешательства.
Сотрудничество нескольких разных специалистов необходимо для успешного управления лечением. Специальный зонд вводится в желудок у новорожденных, у которых есть проблемы с приемом пищи . Однако эта проблема со временем должна исчезнуть, и ребенка можно будет нормально кормить из бутылочки. Если проблемы сохраняются, родители должны проконсультироваться с диетологом. Терапия также включает в себя следующие пункты:
- Лечение ожирения
- Лечение низкого скопления пациентов
- Лечение дефицита половых гормонов
- Профилактика сколиоза
- Овладение мышечной слабостью
- Управление поведенческими расстройствами
ГОРМОНАЛЬНАЯ ТЕРАПИЯ
Частью медикаментозного лечения у пациентов с синдромом Прадера-Вилли является замена гормонов, которые отсутствуют в их организме. Детям дают гормон роста в виде ежедневных подкожных инъекций , которые могут значительно ускорить их рост и оказывают благотворное влияние на неправильный состав тела. Это уменьшает количество подкожного жира, дает ребенку достаточно энергии и даже помогает нормальному развитию лица.
Поскольку у детей с синдромом Прадера-Вилли нарушена гормональная регуляция, также характерна пониженная или полностью отсутствующая активность их половых желез. Терапия, следовательно, состоит из поставки веществ, которые организм не может сделать сам. Использование половых гормонов обычно рекомендуется гинекологом или эндокринологом и должно начинаться в соответствующем возрасте. Девочкам назначают эстроген и прогестерон, а мальчикам — андрогены.
ДИЕТИЧЕСКИЕ РЕКОМЕНДАЦИИ
Мониторинг веса пациентов с синдромом Прадера-Вилли также является одним из наиболее важных поддерживающих методов лечения . Что особенно важно, так это усилия родителей и врачей по предотвращению чрезмерной потери веса , поскольку ребенок не может научиться ограничивать потребление пищи. Люди вокруг него должны быть осведомлены о его диагнозе и уважать его. Другие соответствующие меры включают в себя:
- Акцент на регулярное питание
- Исключение сладких и калорийных продуктов
- Большие порции фруктов и овощей
- Сокращенные порции крахмала
- Храните еду в недоступном для детей месте
- Регулярные упражнения
ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ
В некоторых случаях хирургическое лечение также требуется у пациентов с синдромом Прадера-Вилли. Это могут быть:
- Коррекция сколиоза
- Хирургия нисходящих яичек
- Удаление миндалин и миндалин
- Лечение осложнений ожирения
- Лечение абдоминальных событий
ПОДДЕРЖКА ПАЦИЕНТОВ С PWS
Из-за эмоциональных расстройств и снижения интеллекта поддерживающая психологическая терапия всегда должна быть частью лечения синдрома Прадера-Вилли. За больными должен присматривать детский психолог или психиатр, чтобы помочь им справиться с болезнью и ее проявлениями. Сотрудничество с опытными преподавателями также важно.
С помощью родителей, школы, врачей и друзей люди с PWS могут выполнять те же действия, что и их здоровые сверстники . Они могут учиться, заниматься своими хобби, найти работу и стать независимыми. В Чешской Республике родители и пациенты, страдающие синдромом Прадера-Вилли, ассоциируют гражданскую ассоциацию с синдромом Прадера-Вилли.
Источник