Хронический миелобластный лейкоз код мкб

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Симптомы
- Дифференциальная диагностика
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Хронический миелолейкоз.
Хронический миелолейкоз
Описание
Хронический миелобластный лейкоз (ХМЛ, хронический миелолейкоз, хронический миелоидный лейкоз) — форма лейкоза, которая характеризуется усиленным и нерегулируемым ростом преимущественно миелоидных клеток в костном мозге с их накоплением в крови. ХМЛ — гемопоэтическое клоновое заболевание, основным проявлением которого является пролиферация зрелых гранулоцитов (нейтрофилов, эозинофилов и базофилов) и их предшественников; вариант миелопролиферативного заболевания, ассоциированный с характерной хромосомной транслокацией (филадельфийской хромосомой). В настоящее время основным способом лечения хронического миелолейкоза является таргетная (целевая) терапия иматинибом и другими препаратами, значительно улучшившая показатели выживаемости.
Симптомы
Заболевание часто протекает бессимптомно, выявляясь при рутинном клиническом анализе крови. ХМЛ может проявляться недомоганием, субфебрильной лихорадкой, подагрой, повышенной восприимчивостью к инфекциям, анемией, тромбоцитопенией с кровоточивостью (хотя также может наблюдаться повышенное содержание тромбоцитов). Также отмечается спленомегалия.
ХМЛ часто разделяют на три фазы на основании клинических характеристик и лабораторных данных. В отсутствие лечения, ХМЛ обычно начинается с хронической фазы, в течение нескольких лет прогрессирует в фазу акселерации и, в конечном счёте, развивается бластный криз. Бластный криз — терминальная фаза ХМЛ, клинически подобная острому лейкозу. Одним из факторов прогресии от хронической фазы к бластному кризу является приобретение новых хромосомных аномалий (в дополнение к Филадельфийской хромосоме). Некоторые пациенты к моменту постановки диагноза могут находиться уже в фазе акселерации или в бластном кризе.
Около 85 % пациентов с ХМЛ к моменту постановки диагноза находятся в хронической фазе. В течение этой фазы клинические проявления обычно отсутствуют или имеются «лёгкие» симптомы, такие как недомогание или чувство переполнения живота. Продолжительность хронической фазы различна и зависит от того, насколько рано было диагностировано заболевание, а также от проведённого лечения. В конечном счёте, при отсутствии эффективного лечения, заболевание переходит в фазу акселерации.
Фаза акселерации.
Диагностические критерии перехода в фазу акселерации могут различаться: наиболее широко используются критерии, установленные исследователями онкологического центра Андерсона при Техасском университете, Сокалом с соавторами, а также Всемирной организацией здравоохранения. Критерии ВОЗ, вероятно, наиболее широко распространены, и отличают фазу акселлерации следующим:
• 10-19 % миелобластов в крови или костном мозге.
• >20 % базофилов в крови или костном мозге.
• <100,000 тромбоцитов, вне связи с терапией.
• >1,000,000, вне зависимости от терапии.
• Цитогенетическая эволюция с развитием новых аномалий в добавление к Филадельфийской хромосоме.
• Прогрессирование спленомегалии или увеличение числа лейкоцитов, вне зависимости от терапии.
Фаза акселерации предполагается при наличии любого из указанных критериев. Фаза акселерации указывает на прогрессию заболевания и ожидаемый бластный криз.
Бластный криз.
Бластный криз — финальная стадия развития ХМЛ, протекающая подобно острому лейкозу, с быстрой прогрессией и непродолжительной выживаемостью. Бластный криз диагностируется на основе одного из следующих признаков у пациента с ХМЛ:
• >20 % миелобластов или лимфобластов в крови или костном мозге.
• Крупные группы бластов в костном мозге при биопсии.
• Развитие хлоромы (солидного фокуса лейкемии вне костного мозга).
Базофилия. Высокая температура тела. Изменение аппетита. Изменение веса. Истощение. Лейкоцитоз. Ломота в суставах. Моноцитоз. Нарушение терморегуляции. Недомогание. Нейтрофилез. Общая потливость. Потеря веса. Слабость. Тромбоцитоз. Увеличение СОЭ. Эозинофилия.
Хронический миелолейкоз
Дифференциальная диагностика
ХМЛ следует дифференцировать от лейкемоидной реакции, при которой мазок крови может имееть схожую картину.
Причины
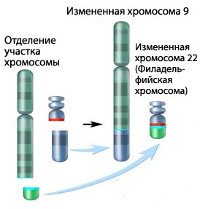
ХМЛ стал первым злокачественным заболеванием с выявленной генетической аномалией, хромосомной транслокацией, которая проявляется патологической Филадельфийской хромосомой. Эта хромосомная патология получила своё название, так как была впервые открыта и описана в 1960 году учёными из Филадельфии, Пенсильвания, США: Питером Ноуеллом (Пенсильванский университет) и Дэвидом Хангерфордом (университет Темпл).
При этой транслокации, части из 9ой и 22ой хромосом меняются местами. В результате, к части BCR-гена из хромосомы 22 прикрепляется ABL-ген из хромосомы 9. Этот аномальный «слившийся» ген генерирует белок p210, или, иногда, p185. Так как abl имеет область, добавляющую фосфатную группу к тирозиновому остатку (тирозин-киназа), продукт аномального гена также является тирозин-киназой.
Белок BCR-ABL взаимодействует с частью клеточного рецептора к ИЛ-3 (CD123-антигеном). Транскрипция BCR-ABL работает непрерывно и не нуждается в активации другими белками. С другой стороны, сам BCR-ABL активирует белковый каскад, контролирующий клеточный цикл, ускоряя деление клеток. Более того, белок BCR-ABL подавляет репарацию ДНК, вызывая неустойчивость генома и делая клетку более восприимчивой к дальнейшим генетическим аномалиям. Активность BCR-ABL — патофизиологическая причина хронического миелолейкоза. С улучшением понимания природы BCR-ABL белка и его действия в качестве тирозин-киназы, была разработана таргетная (целевая) терапия, позволяющая специфически ингибировать активность BCR-ABL белка. Эти ингибиторы тирозин-киназы могут способствовать полной ремиссии ХМЛ, что ещё раз подтверждает ведущую роль bcr-abl в развитии заболевания.
Лечение
Лечение при хроническом миелолейкозе начинают после установления диагноза и обычно проводят амбулаторно.
При отсутствии симптомов хронического миелолейкоза на фоне стабильного лейкоцитоза, не превышающего 40-50- 109/л, применяют гидроксимочевину или бусульфан до достижения содержания лейкоцитов в крови 20*109/л.
По мере прогрессирования хронического миелолейкоза показаны гидроксимочевина (гидрэа, литалир), а-ИФН. При значительной спленомегалии проводят облучение селезёнки.
При выраженной симптоматике хронического миелолейкоза используют комбинации препаратов, применяемых при острых лейкозах: винкристин и преднизолон, цитарабин (цитозар) и даунорубицин (рубомицина гидрохлорид). В начале терминальной стадии иногда эффективен митобронитол (миелобромол).
В настоящее время для терапии хронического миелолейкоза предложен новый препарат — блокатор мутантной тирозинкиназы (р210) — Гливека (STI-571). При бластном кризе ХМЛ и при Ph-позитивных ОЛЛ дозу увеличивают. Применение препарата приводит к полной ремиссии заболевания без эрадикации опухолевого клона.
Трансплантация стволовых клеток крови или красного костного мозга, проводимая больным моложе 50 лет в I стадии заболевания, в 70% случаев приводит к выздоровлению.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: C92.1
МКБ-10 / C00-D48 КЛАСС II Новообразования / C00-C97 Злокачественные новообразования / C81-C96 Злокачественные новообразования лимфоидной, кроветворной и родственных им тканей / C92 Миелоидный лейкоз миелолейкоз
Определение и общие сведения[править]
Хронический миелолейкоз (ХМЛ) — форма лейкоза, которая характеризуется усиленным и нерегулируемым ростом преимущественно миелоидных клеток в костном мозге с их накоплением в крови и ассоциированная с характерной хромосомной транслокацией (филадельфийской хромосомой).
Эпидемиология
В структуре заболеваемости гемобластозами у взрослых ХМЛ занимает 5-е место. Заболеваемость ХМЛ — 1-1,5 случая на 100 000 населения в год. ХМЛ одинаково часто диагностируют среди мужчин и женщин, обычно болеют люди в возрасте 30-50 лет.
В течении ХМЛ выделяют 3 основные фазы (деление на фазы ХМЛ несколько условно):
• хроническую (ХФ);
• фазу акселерации (ФА);
• бластный криз (БК).
Заболевание может быть впервые диагностировано на любом этапе течения.
Этиология и патогенез[править]
Достоверно доказано увеличение частоты развития ХМЛ у лиц, подвергшихся воздействию ионизирующей радиации. Вероятность возникновения ХМЛ повышается с увеличением дозы радиации.
Патогенез
ХМЛ — клональное миелопролиферативное заболевание, возникающее вследствие сбалансированной реципрокной транслокации между хромосомами 9 и 22 [t(9;22)(q34;q11)]. В результате переноса генетического материала возникает характерная филадельфийская (Ph’) хромосома — на 22-й хромосоме образуется химерный ген BCR-ABL, кодирующий белок р210 с высокой тирозинкиназной активностью. Образование тирозинкиназы в гемопоэтических предшественниках приводит к нарушению их нормального функционирования и злокачественному росту.
Клинические проявления[править]
У 85% больных заболевание диагностируется в ХФ, часто начало заболевания протекает бессимптомно, изменения в анализе крови находят случайно [лейкоцитоз (50-100) x 109/л с характерными изменениями лейкоцитарной формулы (количество миелоцитов и нейтрофилов превышает количество метамиелоцитов, возрастает число базофилов и эозинофилов)], нередко уже в дебюте заболевания диагностируют анемию, тромбоцитопению или тромбоцитоз.
В ФА ХМЛ у больных появляется интоксикационный синдром, спленомегалия. В крови нарастает лейкоцитоз, увеличивается число незрелых форм гранулоцитов, в том числе бластных клеток, развивается базофилия и анемия, тромбоцитопения (или, наоборот, тромбоцитоз). Фазу акселерации диагностируют при наличии у больного хотя бы одного из признаков:
• в крови/костном мозге до 15-29% бластных клеток;
• сумма бластов и промиелоцитов в крови и/или костном мозге >30%;
• количество базофилов в крови в крови и/или костном мозге >20%;
• тромбоцитопения <100х109/л, не связанная с химиотерапией;
• увеличение размеров селезенки в процессе лечения;
• дополнительные хромосомные аномалии.
Средняя продолжительность ФА составляет 6-8 мес. У 20-30% больных происходит крайне быстрое прогрессирование заболевания с развитием бластного криза.
Бластный криз (БК)- истинно терминальная стадия заболевания; ее средняя продолжительность 3-6 мес. В фазе бластного криза состояние больных резко ухудшается, возникают интоксикация, лихорадка, оссалгии, повторные инфекции, инфаркты селезенки. В анализе крови — высокое содержание бластных клеток, анемия, глубокая тромбоцитопения с геморрагическим синдромом. Выявляют гепатоспленомегалию, периспленит, экстрамедуллярные очаги кроветворения (миелосаркому).
Хронический миелоидный лейкоз: Диагностика[править]
Лабораторные исследования
Морфологическое исследование крови и пунктата костного мозга — обязательный метод уточнения диагноза, фазы болезни и группы риска. Выявление сочетанной пролиферации грануло-цитарного и мегакариоцитарного ростков, увеличение фиброза в трепанобиоптате костного мозга — факторы неблагоприятного прогноза. Для установления варианта бластного криза ХМЛ необходимо выполнение цитохимического исследования и ИФТ бластных клеток. Учитывая трудности дифференциальной диагностики с хроническими миелопролиферативными заболеваниями (ХМПЗ), лейкемоидными реакциями, диагноз ХМЛ может быть верифицирован только при обнаружении Ph-хромосомы или BCR-ABL-транскрипта.
Для обнаружения Ph’-хромосомы используют стандартное ци-тогенетическое исследование и флюоресцентную in situ гибридизацию (FISH). Высокая чувствительность и специфичность FISH делают этот метод приемлемым и для установления диагноза и мониторинга минимальной остаточной болезни. Основным методом диагностики и мониторинга терапии ХМЛ является количественное определение химерного онкогена BCR-ABL методом ПЦР в реальном времени (real-time ПЦР). Результат real-time ПЦР выражают по отношению к уровню экспрессии контрольного (housekeeping) гена в форме соотношения BCR-ABL/контрольный ген. В качестве контрольного наиболее часто используют гены ABL, BCR, B2M. Для прогнозирования рецидива при мониторинге минимальной остаточной болезни важна динамика изменения уровня транскрипта. Принимая во внимание вариабельность результатов исследований, выполненных в разных лабораториях, с целью стандартизации молекулярных исследований за основу был принят опыт исследования IRIS. За 100% было принято считать средние данные молекулярной оценки у нелеченых больных в исследовании IRIS, а большой молекулярный ответ (БМО) — как 1000-кратную редукцию по сравнению с данными базового значения, т.е. снижение на три десятичных логарифма или соотношение BCR-ABL/ABL, равное 0,1%. Таким образом, основной целью терапии ХМЛ является достижение большого молекулярного ответа, определяемого как тысячекратное снижение уровня BCR-ABL-транскрипта от стартового значения или менее 0,1% по международной шкале.
Дифференциальный диагноз[править]
Хронический миелоидный лейкоз: Лечение[править]
Ранее подходы к терапии больных ХМЛ были преимущественно паллиативными, так как применение с циторедуктивной целью бусульфана или гидроксикарбамида позволяло достигать только временного гематологического ответа у 50-75% взрослых больных. При использовании интерферона-альфа (с/без добавления цитозара) полного цитогенетического ответа (ПЦО) удавалось достичь не более чем у 10% пациентов. Большей эффективности лечения удалось добиться с появлением препаратов семейства ингибиторов тирозинкиназы BCR-ABL, первым представителем которых был иматиниба мезилат (гливек), на протяжении последнего десятилетия являющийся стандартом лечения ХМЛ. Максимальная эффективность препарата показана при его использовании в ранней хронической фазе ХМЛ с возможностью достижения ПЦО и БМО. Полный гематологический ответ (ПГО) развивается у большинства больных через несколько недель приема иматиниба (ИМ), большой цитогенетический ответ (БЦО) — после 3 мес, ПЦО — после 6-9 мес, БМО — после 12-24 мес приема препарата. Доказана не только прогностическая значимость БМО и полного молекулярного ответа (ПМО), фактором благоприятного прогноза считают раннее достижение молекулярной ремиссии: согласно рекомендациям European LeukemiaNet (ELN), достижение БМО к 18-му месяцу считается оптимальным ответом на терапию ингибиторами тирозинкиназ (ИТК1). По данным целого ряда исследователей, аккуратность приема предписанной дозы ИМ определяет вероятность достижения БМО. Рекомендуемые дозы — 400-600 мг в сутки, в ряде случаев повышение дозы до 800 мг в сутки позволяет улучшить результаты терапии. Особую сложность представляет выбор тактики лечения в случае отсутствия оптимального ответа на терапию ИТК. Имеющиеся в настоящее время возможности включают повышение дозы ИМ, применение новых ИТК 2-го поколения (нилотиниб, дазатиниб), участие в клинических исследованиях. Несмотря на оптимистичные результаты терапии ХМЛ ИМ, часть больных не достигают ПЦО или впоследствии его утрачивают, и в ряде случаев возникает устойчивость к препарату. Это послужило причиной создания ИТК 2-го поколения — нилотиниба (Тасигна, Novartis Pharmaceuticals) и дазатинтиба (Спрай-сел; Bristol-Myers Squibb, Princeton, NJ). Нилотиниб — ингибитор BCR-ABL, ранее одобренный для лечения взрослых пациентов с Ph+ ХМЛ в ХФ и ФА при непереносимости или резистентности к терапии ИМ, в настоящий момент одобрен в качестве терапии 1-й линии у больных Ph+ ХМЛ в ХФ. Доза нилотиниба составляет 300 мг 2 раза в сутки в качестве терапии 1-й линии и 400 мг 2 раза в сутки при резистентности или непереносимости ИМ.
Дазатиниб — мультикиназный ингибитор, разрешен к применению для лечения взрослых пациентов с Ph+ ХМЛ во всех фазах при резистентности к ИМ или его непереносимости. Клинические исследования по применению новых ИТК нилотиниба и дазатиниба в ХФ ХМЛ в качестве терапии 1-й линии доказали их преимущество над ИМ в получении ПЦО и БМО, а при применении нилотиниба — также и по снижению риска трансформации в ФА/ БК и увеличению общей выживаемости.
По результатам исследований, при резистентности или непереносимости ИМ нилотиниб и дазатиниб имеют сопоставимые результаты эффективности терапии, однако при различных механизмах действия различаются профили безопасности этих препаратов. Поэтому факторами, определяющими выбор, являются оценка сопутствующих заболеваний и риск развития побочных эффектов. Так, диабет или перенесенный панкреатит могут быть фактором риска при назначении нилотиниба, а использование дазатиниба у больных с застойной сердечной недостаточностью, эпизодами бронхолегочных инфекций и желудочно-кишечных кровотечений в анамнезе может осложняться развитием гидроторакса и кровотечений. Хотя аллогенная трансплантация стволовых кроветворных клеток (АТСКК) является потенциально излечивающей стратегией для больных ХМЛ, благоприятные отдаленные результаты терапии ИМ и многообещающие результаты применения ИТК 2-го поколения изменили роль АТСКК в терапии ХМЛ. Кроме того, альтернативные источники стволовых кроветворных клеток, менее токсичные режимы кондиционирования расширяют возможности для АТСКК. Рекомендации по использованию АТСКК недавно были пересмотрены; показаниями к АТСКК считаются продвинутые фазы заболевания, наличие T315I-мутации, резистентность к терапии ИТК. Оптимальным следует признать проведение АТСКК в ранней фазе заболевания для больных, у которых вовремя выявлена резистентность ко всем имеющимся ИТК. В частности, подходящими кандидатами на АТСКК являются имеющие совместимого донора молодые пациенты, резистентные к ИМ и дополнительно к одному из ИТК 2-го поколения.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Онкология [Электронный ресурс] : Национальное руководство. Краткое издание / под ред. В.И. Чиссова, М.И. Давыдова — М. : ГЭОТАР-Медиа, 2014. — https://www.rosmedlib.ru/book/ISBN9785970431535.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Азацитидин
- Босутиниб
- Гидроксикарбамид
- Дазатиниб
- Даунорубицин
- Иматиниб
- Меркаптопурин
- Митомицин
- Нилотиниб
- Циклофосфамид
- Цитарабин
Источник