Хромосомные синдромы обусловленные геномными мутациями

Хромосомные заболевания вызваны мутациями хромосом (делеции, дупликации, инверсии, транслокации) или изменением числа хромосом (анеуплоидии и полиплоидии). Всего описано около 700 заболеваний, связанных с хромосомной перестройкой. Например, синдром «кошачьего крика».
Кариотип 46 XX или ХУ, 5р-. Хромосомное заболевание — синдром кошачьего крика — объясняется частичной моносомией; оно развивается при делеции (с утратой от трети до половины, реже — полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Изредка отмечается мозаицизм по делении или образование кольцевой хромосомы-5.
При этом синдроме наблюдаются:
- • общее отставание в развитии;
- • низкая масса при рождении и мышечная гипотония;
- • лунообразное лицо с широко расставленными глазами;
- • характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
Кроме того, встречаются врождённые пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, низкое расположение и деформация ушных раковин, кожные складки впереди уха, эпикантус (поперечная кожная складка около внутреннего угла глаза, обычно — двусторонняя). Частота синдрома примерно 1 : 45000. Соотношение полов — мужчины / женщины составляет 1 : 1,3. Интеллект — примерно 20 баллов по шкале интеллекта.
Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьируются по сочетанию врождённых пороков развития органов.
Синдром Дауна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями. Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже — на 14, ещё реже — на 21, 22 и У-хромосому) — 4% случаев, и мозаичный вариант синдрома — 5%.
Английский врач Джон Лэнгдон Даун первым в 1862 году описал и охарактеризовал синдром, впоследствии названный его именем, как форму психического расстройства. Широко известным понятие стало после опубликования им доклада на эту тему в 1866 году. Из-за эпикантуса Даун использовал термин «монголоиды» (синдром же называли «монголизмом»). Представление о синдроме Дауна было очень привязано к расизму вплоть до 1970-х годов. Всемирная организация здравоохранения (ВОЗ) официально убрала название «монголизм» в 1965 году после обращения монгольских делегатов.
Связь между происхождением врождённого синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом.
Первый Международный день человека с синдромом Дауна был проведён 21 марта 2006 года. День и месяц были выбраны в соответствии с номером пары и количеством хромосом.
В XX веке синдром Дауна стал достаточно распространённым. Больные наблюдались, но только малая часть симптомов могла быть купирована. Большинство больных умирали младенцами или детьми. С возникновением евгенического движения в 33-х из 48 американских штатов и в ряде других стран начали программы по принудительной стерилизации лиц с синдромом Дауна и сопоставимыми степенями инвалидности. Это также входило в программу умерщвления Т-4 в нацистской Германии. Судебные проблемы, научные достижения и протесты со стороны общества привели к отменам таких программ в течение десятилетия после окончания Второй Мировой войны.
До середины XX века причины синдрома Дауна оставались неизвестными, однако была известна взаимосвязь между вероятностью рождения ребёнка с синдромом Дауна и возрастом матери (рис. 6.2), также было известно то, что синдрому были подвержены все расы. Синдром Дауна не является редкой патологией — в среднем наблюдается один случай на 700 родов; в данный момент, благодаря пренатальной диагностике, частота рождения детей с синдромом Дауна уменьшилась до 1 : 1100. У обоих полов аномалия встречается с одинаковой частотой.

Рисунок 6.2. Вероятность синдрома Дауна в зависимости от возраста матери
Современные исследования (по состоянию на 2008 год) показали, что синдром Дауна обусловлен также случайными событиями в процессе формирования половых клеток и / или беременности. Поведение родителей и факторы окружающей среды на это никак не влияют. После аварии на Чернобыльской АЭС в январе 1987 года было зарегистрировано необычно большое число случаев синдрома Дауна, однако последующей тенденции к увеличению заболеваемости не наблюдалось.
Трисомия 21-й хромосомы в 95% случаев является причиной возникновения синдрома Дауна, в 88% случаев — из-за нерасхождения материнских гамет и в 8% — мужских. Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, ХХ/47, XX, 21). Данная форма синдрома является, как правило, более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики. По данному типу синдром появляется в 1-2% случаев.
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться за счёт робертсоновских транслокаций в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й [45, XX, der (14; 21) (q10; q10)]).
Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют «семейный синдром Дауна». Это не зависит от возраста матери и показывает скорее равную роль родительских организмов в появлении синдрома Дауна. Данный тип синдрома занимает 2-3% от всех случаев.
Характерные черты, обычно сопутствующие синдрому Дауна:
- • «плоское лицо» — 90%;
- • брахицефалия (аномальное укорочение черепа) — 81%;
- • кожная складка на шее у новорождённых — 81%;
- • эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80%;
- • гиперподвижность суставов — 80%;
- • мышечная гипотония — 80%;
- • плоский затылок — 78%;
- • короткие конечности — 70%;
- • брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) — 70%;
- • катаракта в возрасте старше 8 лет — 66%;
- • открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65%;
- • зубные аномалии — 65%;
- • клинодактилия 5-го пальца (искривлённый мизинец) — 60%;
- • аркообразное нёбо — 58%;
- • плоская переносица — 52%;
- • бороздчатый язык — 50%;
- • поперечная ладонная складка (называемая также «обезьяньей») — 45%;
- • короткая широкая шея — 45%;
- • ВПС (врождённый порок сердца) — 40%;
- • короткий нос — 40%;
- • страбизм (косоглазие) — 29%;
- • деформация грудной клетки, килевидная или воронкообразная — 27%;
- • пигментные пятна по краю радужки — 19%;
- • эписиндром — 8%;
- • стеноз или атрезия двенадцатиперстной кишки — 8%;
- • врождённый лейкоз — 8%.
Точная диагностика возможна на основании анализа крови на кариотип. На основании исключительно внешних признаков постановка диагноза невозможна.
Перспективы развития ребёнка с синдромом Дауна.
Степень проявления задержки умственного и речевого развития зависит как от врождённых факторов, так и от занятий с ребёнком. Дети с синдромом Дауна обучаемы. Занятия с ними по специальным методикам, учитывающим особенности их развития и восприятия, обычно приводят к неплохим результатам.
Наличие дополнительной хромосомы обуславливает появление ряда физиологических особенностей, вследствие которых ребёнок будет медленнее развиваться и несколько позже своих ровесников проходить общие для всех детей этапы развития. Малышу будет труднее учиться, и всё же большинство детей с синдромом Дауна могут научиться ходить, говорить, читать, писать, и вообще делать большую часть того, что умеют делать другие дети.
Синдром Патау (трисомия-13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13. Трисомия-13 впервые описана Эразмусом Бартолином в 1657 году.
Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь. Синдром Патау также был описан для племён с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе. Встречается с частотой 1 : 7000 — 1 : 14000. Соотношение полов при синдроме Патау близко к 1 : 1.
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 грамм). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, короткая шея. У 80% новорождённых встречаются пороки развития сердца. Для синдрома Патау характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95% — до 1 года). Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15% детей) и даже до 10 лет (2-3% детей). Оставшиеся в живых страдают глубокой идиотией.
Синдром Эдвардса (синдром трисомии-18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18-й хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота примерно 1 : 7000. Дети с трисомией-18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7%. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме — для диплоидного набора) в кариотипе зиготы.
Дети с трисомией 18 рождаются с низким, в среднем 2177 грамм, весом. При этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. В 80% случаев наблюдается аномальное развитие стопы: пятка резко выступает, большой палец утолщён и укорочен.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают в возрасте до 3 месяцев, до года доживает лишь 510%. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления синдрома Эдвардса составляет приблизительно 1 : 3000 зачатий и 1 : 6000 рождений живых детей. Хотя женщина в 20 или 30 лет также может родить ребёнка с синдромом Эдвардса, риск рождения больного ребёнка увеличивается с возрастом.
Источник
По изменению генетического материала выделяют три группы мутаций: генные, хромосомные и геномные.
Генными, или точечными (точковыми), называют мутации, возникающие в результате изменения гена, т. е. структуры молекулы ДНК.
При нарушении репликации может произойти изменение последовательности нуклеотидов в каком-нибудь участке ДНК. Это может быть:
- замена нуклеотида;
- вставка нуклеотида;
- выпадение нуклеотида.
Если происходит замена нуклеотида, то результат может быть разный. В некоторых случаях такая мутация не приводит к изменению структуры белка.
Пример:
рассмотрим мутацию ГТТ ЦЦЦ ГГТ → ГТЦ ЦЦЦ ГГТ.
В первом триплете произошла замена тимина на цитозин. Триплеты ГТТ и ГТЦ кодируют глутаминовую кислоту, поэтому никаких изменений в структуре белка данная мутация не вызывает: глу-гли-про → глу-гли-про.
В других случаях замена нуклеотида может изменить порядок аминокислот в молекуле белка и привести к фенотипическим последствиям.
Пример:
мутация ГТТ ЦЦЦ ГГТ → ГТГ ЦЦЦ ГГТ.
В первом триплете произошла замена тимина на гуанин. Триплет ГТТ кодирует глутаминовую кислоту, а триплет ГТГ — гистидин. Значит, первичная структура белка изменяется: глу-гли-про → гис-гли-про. Это может привести к фенотипическим изменениям.
Добавление или выпадение нуклеотидов приводит к сдвигу рамки считывания в рибосоме и к изменению последовательности аминокислот. Синтезируется белок, который отличается своей первичной структурой от исходного. В результате может произойти серьёзное изменение фенотипа.
Пример:
ГТТ ЦЦЦ ГГТ Т → ГТЦ ЦЦГ ГТТ.
Исходный участок ДНК кодирует аминокислотную последовательность глу-гли-про. После выпадения тимина в первом нуклеотиде последовательность аминокислот другая: лиз-глу-глу. Мутагенный ген передаёт к месту синтеза новую информацию, синтезируется другой белок, что может привести к возникновению нового признака.
Генные мутации приводят к таким наследственным заболеваниям, как фенилкетонурия (нарушение обмена веществ) и альбинизм (отсутствие нормальной пигментации).
Хромосомными называют мутации, обусловленные изменением структуры хромосом.
Это может быть:
- утрата (нехватка) — потеря хромосомой своей концевой части;
- делеция — выпадение участка средней части хромосомы;
- дупликация — удвоение фрагмента хромосомы;
- инверсия — поворот участка хромосомы на (180)°;
- транслокация — перенос участка одной хромосомы на другую.
Хромосомные мутации чаще всего возникают при нарушении деления клеток. Их последствия для организма могут быть разными. Наиболее опасны утрата и делеция, так как может быть потеряна информация о жизненно важном белке.
Нарушение структуры хромосом у человека часто приводит к тяжёлым формам умственной отсталости, заболеваниям крови, снижению жизнеспособности организма.
Пример:
потеря небольшой части (21)-й хромосомы вызывает лейкоз.
Хромосомные мутации можно обнаружить с помощью микроскопа. Микроскопирование используется в диагностике наследственных заболеваний.
Геномными называют мутации, обусловленные изменением числа хромосом в кариотипе организма.
Различают полиплоидию и анеуплоидию (гетероплоидию).
Полиплоидия — кратное увеличение гаплоидного набора хромосом.
Возникает при нарушении расхождения хромосом при митозе или мейозе.
В результате хромосомный набор клетки становится триплоидным 3n, тетраплоидным 4n, гексаплоидным 6n и т. д.
Полиплоидия широко используется в селекции растений. Полиплоидные растения, как правило, характеризуются более мощным ростом, большей продуктивностью, жизнеспособностью. Для получения полиплоидных растений используют колхицин, который разрушает нити веретена деления и приводит к образованию полиплоидных геномов.
Диплоидное растение
Полиплоидное растение
Анеуплоидия (гетероплоидия) — некратное изменение числа хромосом 2n±1, 2n±2…
Этот вид мутаций может быть обусловлен избытком или недостатком одной или нескольких хромосом. Причиной гетероплоидии является нарушение расхождения гомологичных хромосом при мейозе. В одну гамету попадают обе гомологичные хромосомы, а в другую — ни одной. Слияние такой гаметы с нормальной и приводит к образованию зиготы с большим или меньшим числом хромосом по сравнению с исходным хромосомным набором.
Различают следующие формы анеуплоидии:
- трисомия (2n+1) — три хромосомы в одной из пар (трисомия по (21)-й паре хромосом у человека — синдром Дауна);
- моносомия (2n−1) — недостаток одной хромосомы (моносомия по X-хромосоме — синдром Шерешевского-Тернера);
- нуллисомия (2n−2) — отсутствие пары гомологичных хромосом (летальная мутация).
Источник
Хромосомные мутации (по-другому их называют аберрациями, перестройками) – это непредсказуемые изменения в структуре хромосом. Чаще всего они вызываются проблемами, возникающими в процессе деления клетки. Воздействие инициирующих факторов среды – это еще одна возможная причина хромосомных мутаций. Давайте же разберемся, какими могут быть проявления такого рода изменений в структуре хромосом и какие последствия они несут для клетки и всего организма.
Мутации. Общие положения
В биологии мутация определяется как стойкое изменение структуры генетического материала. Что значит «стойкое»? Оно передается по наследству потомкам организма, имеющего мутантную ДНК. Происходит это следующим образом. Одна клетка получает неправильную ДНК. Она делится, а две дочерние копируют ее строение полностью, то есть они тоже содержат измененный генетический материал. Далее таких клеток становится все больше, и, если организм переходит к размножению, его потомки получают сходный мутантный генотип.
Мутации обычно не проходят бесследно. Некоторые из них меняют организм настолько, что результатом этих изменений становится летальный исход. Часть из них заставляет организм функционировать по-новому, снижая его способности к адаптации и приводя к серьезным патологиям. И очень малое количество мутаций приносит организму пользу, повышая тем самым его способность адаптироваться к условиям окружающей среды.
Выделяют мутации генные, хромосомные и геномные. Такая классификация основывается на различиях, происходящих в разных структурах генетического материала. Хромосомные мутации, таким образом, затрагивают строение хромосом, генные – последовательность нуклеотидов в генах, а геномные вносят изменения в геном всего организма, прибавляя или отнимая целый набор хромосом.
Поговорим о хромосомных мутациях более подробно.
Какими могут быть хромосомные перестройки?
В зависимости от того, как локализованы происходящие изменения, различают следующие типы хромосомных мутаций.
- Внутрихромосомные – преобразование генетического материала в пределах одной хромосомы.
- Межхромосомные – перестройки, в результате которых две негомологичные хромосомы обмениваются своими участками. Негомологичные хромосомы содержат разные гены и не встречаются в процессе мейоза.
Каждому из этих типов аберраций соответствуют некоторые виды хромосомных мутаций.
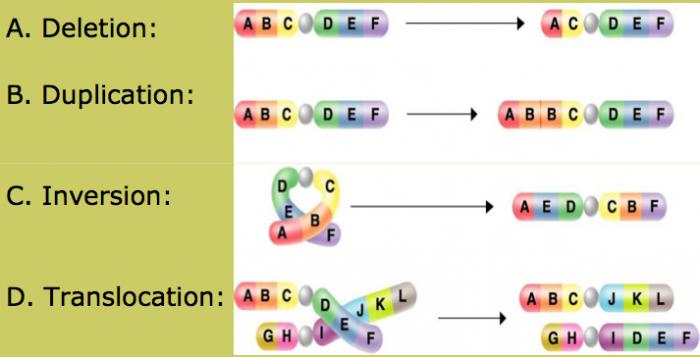
Делеции
Делеция – это отделение или выпадение какого-либо участка хромосомы. Несложно догадаться, что этот тип мутации относится к внутрихромосомным.
Если отделяется крайний участок хромосомы, то делеция называется концевой. Если же происходит выпадение генетического материала ближе к центру хромосомы, такая делеция именуется интерстициальной.
Этот тип мутаций может оказывать влияние на жизнеспособность организма. К примеру, выпадение участка хромосомы, кодирующего определенный ген, обеспечивает человеку невосприимчивость к вирусу иммунодефицита. Эта адаптационная мутация возникла примерно 2000 лет назад и некоторым людям, заболевшим СПИДом, удалось выжить только благодаря тому, что им повезло иметь хромосомы с измененной структурой.
Дупликации
Еще один вид внутрихромосомных мутаций – дупликации. Это копирование участка хромосомы, которое происходит вследствие ошибки при так называемом перекресте, или кроссинговере в процессе деления клетки.
Скопированный таким образом участок может сохранять свое положение, поворачиваться на 180°, или даже повторяться несколько раз, и тогда такая мутация называется амплификацией.
У растений количество генетического материала может увеличиваться именно путем многократных дупликаций. В таком случае обычно меняются способности целого вида к адаптации, а это значит, что такие мутации имеют большое эволюционное значение.
Инверсии
Также относятся к внутрихромосомным мутациям. Инверсия – это поворот определенного участка хромосомы на 180°.
Перевернутая в результате инверсии часть хромосомы может находиться по одну сторону от центромеры (парацентрическая инверсия) или по разные ее стороны (перицентрическая). Центромера – это так называемая область первичной перетяжки хромосомы.
Обычно инверсии не оказывают влияния на внешние признаки организма и не приводят к патологиям. Существует, однако, предположение, что у женщин с инверсией определенного участка девятой хромосомы вероятность выкидыша при беременности возрастает на 30 %.
Транслокации
Транслокация – это перемещение участка одной хромосомы на другую. Эти мутации относятся к типу межхромосомных. Выделяют два вида транслокаций.
- Реципрокные – это обмен двух хромосом определенными участками.
- Робертсоновские – слияние двух хромосом с коротким плечом (акроцентрических). В процессе робертсоновской транслокации короткие участки обеих хромосом утрачиваются.
Реципрокные транслокации приводят у людей к проблемам с деторождением. Иногда такие мутации становятся причиной невынашивания беременности или ведут к появлению на свет детей с врожденными патологиями развития.
Робертсоновские транслокации достаточно часто встречаются у человека. В частности, если транслокация происходит с участием хромосомы 21, у плода развивается синдром Дауна, одна из самых часто регистрируемых врожденных патологий.
Изохромосомы
Изохромосомы – это хромосомы, потерявшие одно плечо, но при этом заменившие его на точную копию другого своего плеча. То есть по сути такой процесс можно считать делецией и инверсией в одном флаконе. В очень редких случаях такие хромосомы имеют две центромеры.
Изохромосомы присутствуют в генотипе женщин, страдающих синдромом Шерешевского – Тернера.
Все описанные выше виды хромосомных мутаций присущи различным живым организмам, в том числе и человеку. Как же они проявляются?
Хромосомные мутации. Примеры
Мутации могут происходить в половых хромосомах и в аутосомах (всех остальных парных хромосомах клетки). Если мутагенез затрагивает половые хромосомы, последствия для организма, как правило, оказываются тяжелыми. Возникают врожденные патологии, которые затрагивают умственное развитие индивида и обычно выражаются в изменениях фенотипа. То есть внешне мутантные организмы отличаются от нормальных.
Геномные и хромосомные мутации чаще возникают у растений. Однако встречаются они и у животных, и у человека. Хромосомные мутации, примеры которых мы рассмотрим ниже, проявляются в возникновении тяжелых наследственных патологий. Это синдром Вольфа-Хиршхорна, синдром «кошачьего крика», болезнь частичной трисомии по короткому плечу хромосомы 9, а также некоторые другие.
Синдром «кошачьего крика»
Это заболевание было открыто в 1963 году. Возникает оно из-за частичной моносомии по короткому плечу хромосомы 5, обусловленной делецией. Один из 45 000 детей рождается с этим синдромом.
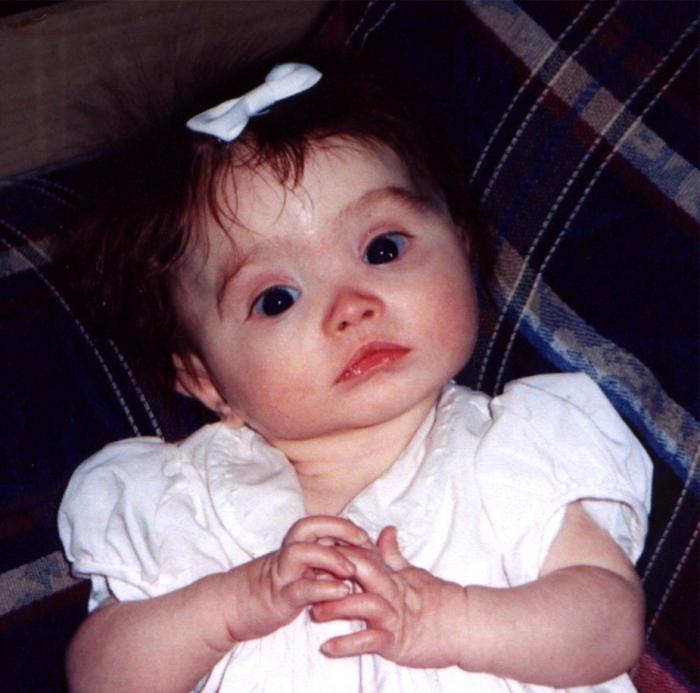
Почему это заболевание получило такое название? Дети, страдающие этой болезнью, имеют характерный плач, который напоминает кошачье мяуканье.
При делеции короткого плеча пятой хромосомы могут утрачиваться разные его участки. Клинические проявления заболевания напрямую зависят от того, какие гены были утеряны в ходе этой мутации.
Строение гортани изменяется у всех больных, а значит «кошачий крик» характерен всем без исключения. У большей части страдающих этим синдромом отмечается изменение строения черепа: уменьшение мозгового отдела, лунообразная форма лица. Ушные раковины при синдроме «кошачьего крика» обычно расположены низко. Иногда у больных отмечаются врожденные патологии сердца или других органов. Характерным признаком также становится умственная отсталость.
Обычно больные с этим синдромом умирают в раннем детстве, лишь 10% из них доживает до десятилетнего возраста. Однако зафиксированы и случаи долгожительства при синдроме «кошачьего крика» – до 50 лет.
Синдром Вольфа-Хиршхорна
Этот синдром встречается значительно реже – 1 случай на 100 000 рождений. Обусловлен он делецией одного из сегментов короткого плеча четвертой хромосомы.
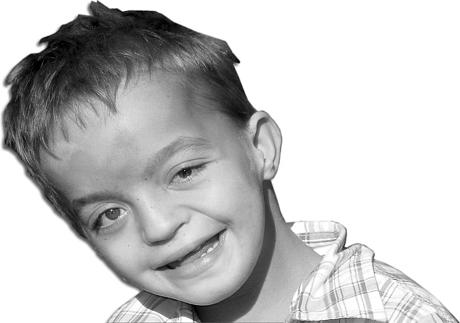
Проявления этого заболевания разнообразны: задержка развития физической и психической сферы, микроцефалия, характерная клювовидная форма носа, косоглазие, расщелины неба или верхней губы, маленький рот, пороки внутренних органов.
Как и многие другие хромосомные мутации человека, болезнь Вольфа-Хиршхорна относится к категории полулетальных. Это значит, что жизнеспособность организма при такой болезни существенно снижена. Дети с диагностированным синдромом Вольфа-Хиршхорна обычно не доживают до 1 года, однако зафиксирован один случай, когда больной прожил 26 лет.
Синдром частичной трисомии по короткому плечу хромосомы 9
Возникает это заболевание по причине несбалансированных дупликаций в девятой хромосоме, в результате чего генетического материала в этой хромосоме становится больше. Всего известно более 200 случаев таких мутаций у человека.
Клиническая картина описывается задержкой физического развития, легкой умственной отсталостью, характерным выражением лица. Пороки сердца обнаруживаются у четвертой части всех больных.
При синдроме частичной трисомии короткого плеча хромосомы 9 прогноз все же относительно благоприятный: большая часть больных доживают до пожилого возраста.

Другие синдромы
Иногда даже на очень маленьких участках ДНК происходят хромосомные мутации. Болезни в таких случаях обычно обусловлены дупликациями или делециями, и их называют соответственно микродупликационными или микроделеционными.
Самым распространенным таким синдромом считается болезнь Прадера-Вилли. Возникает она из-за микроделеции участка хромосомы 15. Что интересно, эта хромосома должна быть обязательно получена организмом от отца. В результате микроделеции затронутыми оказываются 12 генов. У больных с этим синдромом отмечаются умственная отсталость, ожирение, а также у них обычно маленькие стопы и кисти рук.
Еще одним примером таких хромосомных болезней может служить синдром Сотоса. Происходит микроделеция на участке длинного плеча хромосомы 5. Клиническая картина этого наследственного заболевания характеризуется быстрым ростом, увеличением в размерах кистей рук и стоп, наличием выпуклого лба, некоторой задержкой психического развития. Частота встречаемости этого синдрома не установлена.
Хромосомные мутации, точнее, микроделеции на участках 13 и 15 хромосом, вызывают соответственно опухоль Вильмса и ретинбластому. Опухоль Вильмса – это рак почек, который возникает преимущественно у детей. Ретинобластома – это злокачественная опухоль сетчатки, которая также встречается у детей. Эти заболевания лечатся, если диагностика их проведена на ранних стадиях. В некоторых случаях врачи прибегают к оеративному вмешательству.

Современная медицина избавляет от многих болезней, но вылечить или хотя бы предотвратить хромосомные мутации пока нельзя. Их можно только выявить в начале внутриутробного развития плода. Однако генная инженерия не стоит на месте. Быть может, в скором времени способ предотвращения болезней, вызываемых хромосомными мутациями, будет найден.
Источник