Гликогеновая болезнь код мкб

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
E74,0 Болезни накопления гликогена.
E74.0 Болезни накопления гликогена
Синонимы диагноза
Болезни накопления гликогена, гликогенозы, помпе болезнь.
Описание
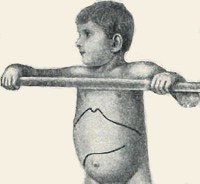
Болезнь Помпе. Редкая наследственная патология, одна из форм лизосомных болезней накопления, характеризующаяся нарушением процессов расщепления гликогена в нервных и мышечных клетках (скелетные мышцы, миокард). Симптомы заболевания довольно вариабельны по времени своего проявления и выраженности у разных больных, традиционно наблюдается прогрессирующая мышечная слабость, при некоторых формах – кардиомегалия с дилятационной кардиомиопатией. Диагностика болезни Помпе производится на основании данных наследственного анамнеза, гистологического и гистохимического изучения мышечных тканей, биохимического анализа крови, а также генетических исследований. Лечение в настоящий момент может производиться с помощью фермент-заместительной терапии, однако эффективность этой методики неодинакова у разных пациентов.
E74.0 Болезни накопления гликогена
Дополнительные факты
Болезнь Помпе (гликогеноз 2-го типа, недостаточность кислой альфа-глюкозидазы) – наследственное заболевание, при котором из-за нарушения процессов обмена гликогена происходит повреждение нервных и мышечных тканей. Впервые было описано в 1932 году голландским ученым И. Помпе, с тех пор официально зарегистрировано более 50 случаев патологии. Болезнь Помпе с равной степенью вероятности поражает как мужчин, так и женщин, встречаемость колеблется от 1:60000 (взрослая форма) до 1:140000 (ранняя, или инфантильная форма). Является одной из немногих лизосомных болезней накопления, в отношении которой было разработано эффективное специфическое лечение, одобренное в США в 2006 году и в России – в 2013 г. Однако стоимость этиотропной терапии болезни Помпе крайне высока и составляет несколько сотен тысяч долларов в год. Смертность в случае отсутствия лечения зависит от формы патологии – детская инфантильная форма часто приводит к летальному исходу на 1-2-м году жизни ребенка, при типе болезни с отсроченным началом нарастание симптомов идет намного медленнее.
Причины
Болезнь Помпе является классическим гликогенозом, при ней в тканях скелетных мышц, миокарда и отчасти нервной системы формируются отложения гликогена по причине невозможности его расщепления. Это происходит в результате мутации гена GAA, расположенного на 17-й хромосоме – он кодирует последовательность кислой альфа-1,4-глюкозидазы или мальтазы. Это один из ключевых ферментов лизосом, участвующий в расщеплении молекулы гликогена на более простые отрезки, которые, в конечном итоге, деградируют до глюкозы, вступающей в энергетический обмен клетки. Так как гликоген является важным депо энергии для таких структур, как скелетные мышцы, миокард, печень и нервная ткань, проявления болезни Помпе сводятся именно к патологическим изменениям данных органов.
В результате подобных изменений сначала возникает дефицит энергии в клетках – потребности тканей в глюкозе покрываются только за счет ее поступления из крови. Кроме того, в лизосомах при болезни Помпе начинает накапливаться гликоген, формируя крупные включения в виде вакуолей, в дальнейшем приводя к дистрофии и повреждению клеток. Наследование дефектных вариантов гена GAA происходит по аутосомно-рецессивному типу. Наличие нескольких форм заболевания предположительно объясняется разными типами мутаций вышеуказанного гена. Возможно, при некоторых дефектах происходит не полное исчезновение, а лишь снижение активности кислой альфа-1,4-глюкозидазы, что и приводит к более позднему развитию болезни Помпе и медленному прогрессированию заболевания. Определение формы патологии играет важную роль для составления ее прогноза и схемы лечения.
Классификация
На сегодняшний день специалисты выделяют несколько основных форм болезни Помпе, основное различие между которыми заключается в сроках начала заболевания и выраженности симптомов. В большинстве случаев, с гликогенозом 2-го типа сталкиваются врачи-педиатры, однако имеется тип заболевания, выявляемый у взрослых.
• Ранняя инфантильная форма. Такая разновидность болезни Помпе считается наиболее тяжелой. Выявляется еще на первых месяцах жизни, симптомы миопатии, поражения печени (гепатомегалия) и сердца (кардиомиопатия) достаточно быстро прогрессируют. Обычно больные с ранней инфантильной формой болезни Помпе умирают от сердечной или дыхательной недостаточности в возрасте до года.
• Поздняя инфантильная форма. Первые симптомы возникают в возрасте 1 — 3 года, скорость их прогрессирования также намного медленнее. При данном типе болезни Помпе наиболее выражены поражения миокарда, смерть наступает к подростковому возрасту от сердечной недостаточности.
• Ювенильная форма болезни Помпе развивается в возрасте 6-10 лет. Так же, как и в предыдущем варианте, основным органом-мишенью болезни становится сердце, смерть от нарастающей сердечной недостаточности наступает к 20 годам.
• Взрослая форма болезни Помпе. Манифестация симптомов происходит в 20 — 40 лет. Ведущим симптомом является медленно прогрессирующая миопатия, поражения печени практически никогда не регистрируются, в некоторых случаях возможны незначительные нарушения миокарда. При этом типе болезни Помпе больные во многих случаях доживают до старости, лишь иногда возможен более ранний летальный исход из-за дыхательной или сердечной недостаточности.
Методами современной генетики на сегодняшний момент не определена взаимосвязь между отдельными типами мутаций гена GAA и формами болезни Помпе. Возможно, причина такой вариабельности проявлений лежит совсем в другом – в литературе описаны семейные случаи заболевания, когда у родственников регистрировались различные формы патологии. Изучение закономерностей, приводящих к развитию болезни Помпе определенного типа, сильно осложняется относительной редкостью данного синдрома.
Симптомы
Проявления болезни Помпе довольно сильно отличаются при различных формах заболевания. Ранний инфантильный тип характеризуется выраженной мышечной слабостью младенца, снижением его двигательной активности, плаксивостью. В педиатрии при осмотре такого больного часто выявляется задержка психомоторного развития, различная степень увеличения печени, пальпация иногда выявляет гипертрофию мышц, которые, однако, при этом довольно слабые. При дальнейшем развитии болезни Помпе возникают проблемы с кормлением из-за слабости сосательной мускулатуры, выявляется дисфагия и, как итог всего этого – гипотрофия. Нарастающая кардиомиопатия и слабость дыхательной мускулатуры со временем приводят к смерти ребенка.
Поздняя инфантильная и ювенильная формы болезни Помпе протекают практически одинаково, различается только срок появления симптомов патологии. Как правило, выявляется мышечная слабость, признаки кардиомиопатии. Со временем начинает формироваться выраженная дистрофия скелетной мускулатуры, кардиомегалия, на этом фоне начинают увеличиваться печень и селезенка. Длительность течения этих форм болезни Помпе составляет около 10-12 лет, после чего, при отсутствии лечения, наступает летальный исход из-за декомпенсированной сердечной недостаточности. Косвенным симптомом будет являться большая частота простудных заболеваний с легочными осложнениями, ночное апноэ, головные боли по утрам.
Диагностика
Выявление болезни Помпе можно производить многочисленными клиническими методиками – биохимическим анализом крови, изучением биоптата мышц, культур фибробластов или лейкоцитов больного, традиционными исследованиями (осмотр, ЭКГ, ЭхоКГ). При осмотре часто обнаруживается слабость и дистрофия мышц, при вовлечении в патологический процесс внутренних органов – гепато- и спленомегалия. На электрокардиограмме регистрируется укорочение интервала PQ, расширение комплекса QRS, обусловленного увеличением размеров миокарда. По этой же причине увеличивается длительность фазы реполяризации желудочков, что проявляется инверсией зубца T. Эхографическое исследование сердца показывает резкое увеличение его размеров за счет значительного утолщения стенок желудочков. При взрослой форме болезни Помпе вышеуказанные изменения миокарда могут не выявляться.
Биохимический анализ крови позволяет обнаружить как специфические, так и косвенные признаки заболевания. Специфическим исследованием будет определение активности кислой альфа-1,4-глюкозидазы в плазме крови, которая при болезни Помпе будет резко снижена. Косвенным указанием на наличие патологии является резкое повышение активности креатинфосфокиназы, обусловленное поражением мышечной ткани. Гистологическое исследование биоптата мышц выявляет в миоцитах многочисленные включения гликогена, часто придающие им вид «пенистых клеток». Гистохимическое изучение при болезни Помпе выявляет резкое снижение активности кислой альфа-1,4-глюкозидазы в мышечной ткани, фибробластах и лейкоцитах.
Врачом-генетиком может быть проведено генетическое определение болезни Помпе – оно производится методом прямого секвенирования последовательности гена GAA с целью выявления дефектных участков. Кроме того, может помочь в диагностике заболевания и составление наследственного анамнеза. Генетическая диагностика болезни Помпе включает в себя секвенирование GAA и у фенотипически здоровых родственников больного с целью выявления носительства патологического гена.
Лечение
На сегодняшний день единственным методом специфического лечения болезни Помпе является фермент-заместительная терапия с целью восполнения дефицита кислой альфа-1,4-глюкозидазы. Для этого используют препарат альфа алглюкозидазы производства США. Стоимость этого лечения крайне высока (годовой курс стоит 100-400 тысяч долларов), однако его эффективность неодинакова у разных больных. Других способов лечения болезни Помпе в настоящее время не существует.
Прогноз
Без лечения при инфантильных и ювенильной формах заболевания прогноз неблагоприятный, у взрослого типа – неопределенный. Профилактика возможна только путем своевременного выявления носительства болезни Помпе (в случае наличия патологии у кровных родственников) и последующей генетической пренатальной диагностики.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
- Описание
- Лечение
Краткое описание
Гликогенозы — группа наследственных заболеваний, вызванных недостаточностью одного или нескольких ферментов, вовлечённых в синтез и распад гликогена, и характеризующихся накоплением патологических количеств или типов гликогена в тканях. Симптоматика возникает вследствие накопления гликогена или его промежуточных метаболитов или из — за недостатка конечных продуктов распада гликогена, особенно глюкозы. Гликоген и некоторые из промежуточных метаболитов, депонированных в тканях, могут быть обнаружены при МРТ. Различия в степени тяжести и возрасте начала клинических проявлений вызваны вовлечением различных изоферментов или других компонентов повреждённых ферментных систем. Частота всех форм болезней накопления гликогена — 1:40 000 населения.
Код по международной классификации болезней МКБ-10:
- E74.0 Болезни накопления гликогена
Генетическая классификация и клиническая картина
• Гликогеноз типа 0 (*240600, 12p12.2, ген GYS2 [138571], r) — недостаточность гликоген синтетазы (КФ 2.4.1.11) печени. Клиническая картина: гипогликемия и гиперкетонемия натощак, судороги, гипергликемия и гиперлактатемия после приёма пищи.
• Гликогеноз типа I(a) (232200, 17q21, Â) — недостаточность глюкозо — 6 — фосфатазы (КФ 3.1.3.9), приводящая к избыточному накоплению гликогена нормальной химической структуры (особенно в печени и почках). Наблюдают значительно чаще других гликогенозов. Клиническая картина: гипогликемия, артериальная гипертензия, задержка роста, позднее половое созревание, увеличение живота, аденомы печени, печёночноклеточная карцинома, гепатобластома, увеличение печени, хронический панкреатит, ксантомы, паукообразные гемангиомы, подагрические тофусы, протеинурия, гематурия, почечная недостаточность, центральный сегментарный гломерулосклероз, мочекислые камни почек, подагрический артрит, геморрагический диатез, лёгочная артериальная гипертензия. Лабораторные данные: недостаточность глюкозо — 6 — фосфатазы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. Синонимы: фон Гирке болезнь, нефромегалический гликогеноз, фон Гирке ван Кревельда синдром, фон Гирке ван Кревельда болезнь.
• Гликогеноз типа Ib (232220, r) — мутации гена транспортёра глюкозо 6 фосфата (*602671, 11q23.3). Клиническая картина: диарея, плохой аппетит, болезнь Крона, хронические остеомиелиты, перианальные абсцессы, нейтропения, гипохромная анемия, тромбоцитопения, вторичный амилоидоз, протеинурия, гиперлипидемия.
• Гликогеноз типа Ic (232240, r) — дефект транспортёра глюкозо 6 фосфата (*602671, 11q23.3). Клиническая картина: гипогликемия, артериальная гипертензия, протеинурия, гематурия, почечная недостаточность, центральный гломерулосклероз, задержка роста и полового созревания, опухоли печени (аденомы, печёночноклеточная карцинома, гепатобластома), увеличение печени, хронический панкреатит, ксантома, ангиома кожи, подагрические тофусы, подагрический артрит, нормальные функции лейкоцитов, лёгочная артериальная гипертензия. Лабораторные данные: дефицит фосфат пирофосфат транслоказы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. ЛС (аллопуринол) следует назначать с осторожностью. Течение обычно хроническое прогрессирующее.
• Гликогеноз типа IIa (153360 — недостаточность лизосомной a — 1,4 глюкозидазы, приводящий к избыточному накоплению гликогена нормальной химической структуры в сердце, скелетных мышцах, печени, мозге. Клиническая картина: кардиомиопатия, кардиомегалия, артериальная гипотензия, миотония, мышечная слабость, утомляемость, увеличенный язык, смерть на первом году жизни, дыхательная недостаточность, одышка, аневризмы мозговых артерий. Синоним: Помпе болезнь.
• Гликогеноз типа IIb (300257, дефект ассоциированного с лизосомами мембранного белка LAMP2, r). Клиническая картина: слабость проксимальных мышц, гипертрофическая кардиомиопатия, сердечная недостаточность, АВ — блокада, умственная отсталость. Лабораторные данные: накопление гликогена в лизосомах; активность кислой мальтазы нормальная. Синонимы: болезнь Антополя, болезнь Данона (300257, дефект лизосомного белка LAMP2).
• Гликогеноз типа III (*232400, 1p21, ген AGL, GDE, r) — недостаточность амило — 1,6 — глюкозидазы, приводящая к накоплению гликогена ненормальной структуры с короткими внешними цепями в печени и мышцах. Клиническая картина: миопатия, увеличение печени, гипогликемия, кетоацидоз, мышечная слабость с атрофией мышц, «ангельское» лицо, склонность к кровотечениям (в т.ч. носовым), гипертрофия желудочков на ЭКГ. Синонимы: Кори болезнь, Форбса болезнь, лимитдекстриноз.
• Гликогеноз типа IV (*232500, 3p12, ген GBE1, r) — недостаточность 1,4 — a — глюкан ветвящего фермента (КФ 2.4.1.18), приводящая к накоплению гликогена ненормальной структуры с длинными цепями в печени, почках, мышцах и других тканях. Клиническая картина: цирроз печени, портальная гипертензия, печёночная недостаточность, гипогликемия, кардиомиопатия, сердечная недостаточность, миопатия, тазово — плечевая мышечная дистрофия, гиперлордоз позвоночника, походка вразвалку, слабость проксимальных мышц конечностей, смерть до 4 — летнего возраста. Синонимы: болезнь Андерсен, амилопектиноз.
• Гликогеноз типа V (*232600, 11q13, ген PYGM, r) — дефект амилофосфорилазы (КФ 2.4.1.1), вызывающий накопление гликогена нормальной химической структуры в мышцах. Клиническая картина: слабость и атрофия скелетных мышц, мышечные боли при нагрузке, миоглобинурия. Синонимы: МакАрдла–Шмида–Пирсона болезнь, миофосфорилазная недостаточность, МакАрдла болезнь.
• Гликогеноз типа VI (*232700, 14q21–q22, ген PYGL, r) — недостаточность амилофосфорилазы (КФ 2.4.1.1), приводящая к накоплению гликогена нормальной структуры в гепатоцитах и лейкоцитах. Клиническая картина: увеличение печени, кетоз, гипогликемия, задержка роста. Синонимы: Гирса болезнь, гепатофосфорилазная недостаточность.
• Гликогеноз типа VII (*232800, 12q13.3, ген PFKM, r) — миопатии и увеличение печени, обусловленные недостаточностью 6 фосфофруктокиназы (КФ 2.7.1.11). Клиническая картина: миопатия, увеличение печени, слабость мышц, крампи, гемолиз, лёгкая полицитемия, ретикулоцитоз, умеренная желтуха, желчнокаменная болезнь. Лабораторные данные: недостаточность фосфофруктокиназы мышц, миоглобинурия, гиперурикемия. Синонимы: гепатофосфоглюкомутазная недостаточность, Томсона болезнь.
• Гликогеноз типа VIII (*261750, Xp22.2 p22.1, ген PHKA2, PHK; *311870, Xq12 q13, ген PHKA1, À) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в мышцах. Клинические и биохимические расстройства исчезают с возрастом, у большинства взрослых пациентов заболевание протекает бессимптомно. Клиническая картина: увеличение печени, задержка роста, почечный канальцевый ацидоз. Лабораторные данные: недостаточность печёночной киназы фосфорилазы (PHK); мышечная киназа фосфорилазы в пределах нормы; повышенное содержание глутамат — пируват и глутамат оксало — ацетат трансаминаз; гиперхолестеринемия; гипертриглицеридемия; кетонемия на фоне голодания; гиперлактацидемия или гиперурикемия отсутствует. Синонимы: Таруи болезнь, недостаточность киназы фосфорилазы печени и мышц. Примечание: некоторые субъединицы фермента имеют локусы в 16p12.1 p11.2.
• Гликогеноз типа VIIIb (261740, r) — крайне редкая форма недостаточности фосфофруктокиназы (КФ 2.7.1.38), ограниченная мышцей сердца.
• Гликогеноз типа VIIIc (*261750, r) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в печени и мышцах. Клиническая картина: увеличение печени, диарея, задержка роста, гипотония мышц, умеренная слабость. Лабораторные данные: недостаточность киназы фосфорилазы в печени и мышцах с накоплением гликогена.
Лечение
ЛЕЧЕНИЕ • При типах 0, I и III — предотвращение гипогликемии и молочного ацидоза назначением дробных доз углеводов, что позволяет поддержать нормальные уровни глюкозы крови, предупредить развитие молочного ацидоза, гиперурикемии и гиперлипидемии. Кроме того, используют непрерывную подачу высокомолекулярных декстранов через эндоназальный зонд. Аллопуринол назначают для профилактики подагры и уратных камней почек • Ограничение анаэробной нагрузки уменьшает мышечные симптомы типов V и VII • При типе VIII рекомендуют ограничение физической нагрузки и обильное питьё • Эффективных методов лечения других типов нет.
МКБ-10 • E74.0 Болезни накопления гликогена
Источник