Генетические заболевания печени по синдромам

Этот сайт сделан экспертами: токсикологами, наркологами, гепатологами. Строго научно. Проверено экспериментально.

Автор этой статьи, эксперт: Гастроэнтеролог-гепатолог Екатерина Кашух
Вкратце: Существуют разные генетические заболевания печени: одни приводят к смерти ещё в младенчестве, а с другими можно прожить долгую жизнь с минимальными ограничениями.
Какие бывают генетически обусловленные заболевания печени
Многообразие функций печени создаёт предпосылки для большого количества путей её поражения. Заболевания данного органа можно разделить на приобретённые и генетически обусловленные.
Среди генетически обусловленных болезней печени выделяют:
- Наследуемые по аутосомно-доминантному типу. В этом случае при наличии гена у одного из родителей он обязательно проявит себя в виде заболевания у ребёнка.
- Наследуемые по аутосомно-рецессивному типу. При таком наследовании для возникновения болезни печени необходимо, чтобы патологический ген был у обоих родителей.
Генетически обусловленными заболеваниями называют группу гепатозов, которые развиваются при нарушении обмена разных соединений, например:
- аминокислот и белков (болезнь цистиноз);
- жиров (липидоз);
- углеводов (гликогеноз);
- металлов (гемохроматоз, болезнь Вильсона и Коновалова);
- пигментов (порфирии, доброкачественные гипербилирубинемии).
Печень участвует во всех этих видах обмена, однако наряду с поражением её ткани отмечаются дистрофические изменения и других органов.
Дистрофией называется расстройство клеточного и тканевого метаболизма, приводящее к структурным изменениям в органе. В основе дистрофий лежат ферментопатии (несостоятельность определенных ферментов).
Дистрофия приводит к гибели гепатоцитов (рабочих клеток печени) и активации фиброза (накопление соединительнотканных волокон). В ответ развиваются процессы регенерации, образуются узлы между фиброзными участками, орган старается компенсировать потерянные участки функционально активной паренхимы.
Нарушение обмена билирубина
Достаточно большую группу заболеваний печени, обусловленных дефектами в работе генов, составляют доброкачественные гипербилирубинемии. Причина их развития связана с дефектом фермента, участвующего в обмене билирубина. Происходит нарушение его биохимического преобразования, что клинически сопровождается развитием желтухи.
К этой группе заболеваний относятся несколько синдромов, названных по имени описавших их учёных. Основные характеристики доброкачественных гипербилирубинемий, используемые для их дифференциации, представлены в таблице.
| название синдрома (и механизмы развития) | клинические проявления | методы лечения |
|---|---|---|
| Жильбера (недостаточная активность фермента уридиндифосфатглюкуронилтрансферазы (УДФГТ) печени, приводящая к снижению захвата билирубина гепатоцитами) | чаще у лиц мужского пола, дебютирует в возрасте 10-25 лет. Проявляется периодически возникающими эпизодами желтухи, которые провоцируются стрессом, алкоголем, физическими нагрузками, сопутствующими заболеваниями. Разрешается самостоятельно, прогноз для жизни благоприятный | ранее применялся фенобарбитал, в настоящее время запрещён. Не требует специфического лечения, не является жизнеугрожающим состоянием |
| Криглера-Найяра 1 тип (полное отсутствие фермента УДФГТ) | проявляется через несколько часов после рождения, высокий билирубин в крови становится причиной поражения нервной системы — развития печёночной энцефалопатии | проводится фототерапия, обменные гемотрансфузии. Больные обычно умирают от поражения головного мозга в течение первых 2 лет жизни. Прогноз улучшается при трансплантации печени |
| Криглера-Найяра 2 тип (фермент УДФГТ есть, но активность его снижена) | манифестирует через несколько месяцев (иногда лет) после рождения | используется фенобарбитал, сеансы фототерапии. Прогноз для жизни более благоприятный |
| Дабина и Джонсона (нарушена транспортировка билирубина из гепатоцитов в желчь) | проявляется в молодом возрасте (23-28 лет), эпизоды желтухи сопровождаются болями в животе, провоцируются инфекциями, беременностью, приёмом алкоголя, оральных контрацептивов | терапия не разработана, рекомендуется исключить приём алкоголя, придерживаться диеты, прогноз для жизни благоприятный |
| Ротора (нарушена доставка и конъюгация билирубина) | наблюдается чаще у подростков мужского пола, проявляется умеренной желтухой, болями в животе, диспепсическими явлениями, незначительным повышением температуры тела | лечение не разработано, прогноз для жизни благоприятный |
Среди пигментных гепатозов самый неблагоприятный прогноз имеет синдром Криглера-Найяра 1 типа.
Профилактика всех гепатозов является неспецифической и сводится к:
- отказу от вредных привычек и бесконтрольного приёма лекарств,
- правильному питанию,
- исключению чрезмерных нагрузок и стрессов.
К этой категории наследственных болезней относятся:
- врождённый гемохроматоз
- и болезнь Вильсона-Коновалова.
Обе патологии наследуются по аутосомно-рецессивному типу. То есть у ребёнка проявляется заболевание только в том случае, если у обоих родителей соответствующий ген дефектный.
Печень повреждается в результате окислительных процессов, индуцированных ионами железа при гемохроматозе и меди при синдроме Вильсона-Коновалова. В первом случае патологический ген находится в 6 хромосоме, во втором случае — в 13-й.
Гемохроматоз
При гемохроматозе нарушено всасывание железа из-за патологии тонкой кишки. Такая ситуация приводит к накоплению этого металла в печени и других органах: сердце, поджелудочной железе и др.
Заболевание может проявлять себя во второй половине жизни, у мужчин раньше, чем у женщин, в связи с регулярной потерей крови (и железа, соответственно) женщинами при менструации.
Среди проявлений гемохроматоза могут присутствовать:
- гиперпигментация кожи (коричнево-серый цвет), слизистых оболочек и сетчатки;
- общая слабость
- сердечная недостаточность;
- сахарный диабет;
- отёк и болезненность суставов.
Диагноз устанавливается при обнаружении повышенного уровня железа крови и изменения показателей его обмена (ферритин, трансферрин). Результаты анализа крови дополняются ультразвуковым или магнитно-резонансным исследованием печени, биопсии печени и кожи. В настоящее время наиболее точно можно выявить наличие гемохроматоза по анализу крови на наличие мутаций гена HFE.
Оптимальным лечением гемохроматоза считаются регулярные кровопускания. Также используются препараты, связывающие железо и препятствующие его отложению во внутренних органах. При развитии цирроза возможна трансплантация печени.
Важно! При хроническом злоупотреблении алкоголем, некоторых видах анемии и повторных переливаниях крови развивается вторичный гемохроматоз, то есть повышенное отложение железа в органах. Эти состояния необходимо отличать от генетически обусловленного нарушения обмена железа.
Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова представляет собой редкое аутосомно-рецессивное заболевание, при котором в органах и тканях происходит избыточное накопление меди (так же как при гемохроматозе избыточно накапливается железо).
Медь в повышенной концентрации оказывает токсическое действие на клетки печени, что приводит к хроническому гепатиту с вероятным развитием цирроза печени. Накопление меди пагубно влияет не только на печень, но и на головной мозг, почки, кости, органы зрения, суставы.
Вследствие влияния на многие органы болезнь Вильсона-Коновалова может иметь множество различных проявлений и «клинических масок», поэтому заподозрить её на начальных этапах бывает очень сложно.
Заболевание начинает проявляться к подростковому возрасту, однако может быть бессимптомным до зрелого возраста. Кроме изменений в биохимическом анализе крови, характерных для хронического гепатита, у пациентов выявляют:
- неврологические и психические расстройства,
- офтальмологические проблемы (в т. ч. кольцо Кайзера-Флейшера — желтовато-зелёная или зеленовато-коричневая пигментация по периферии роговой оболочки глаза),
- нарушение функции почек,
- анемию и др.
Диагностика проводится при наличии признаков поражения печени, нервной системы, обнаружения колец Кайзера-Флейшера в сочетании с биохимическими признаками избыточного накопления меди в организме (анализ на церулоплазмин крови, суточную экскрецию меди с мочой), а также с характерной гистологической картиной при биопсии печени. Используются ультразвуковые и магнитно-резонансные методы визуализации изменений в печени.
Для лечения болезни Вильсона-Коновалова могут применяться препараты, снижающие усвоение меди в органах и тканях, наряду с диетой, ограничивающей избыточное поступление данного металла в организм. При развитии цирроза показана трансплантация печени.
Дефицит лизосомной кислой липазы
Дефицит лизосомной кислой липазы (ДЛКЛ) — это редкое наследственное заболевание, при котором из-за недостатка фермента лизосомной кислой липазы (ЛКЛ) происходит накопление в клетках лизосом, и они перестают функционировать. Этот фермент необходим человеку для процесса обмена жиров в печени и других органах.
Выделяют две формы ДЛКЛ:
- Болезнь Вольмана (БВ). Выявляется уже в младенчестве и имеет менее благоприятный прогноз. Диагноз ДЛКЛ может быть подтверждён снижением активности фермента ЛКЛ в культуре фибробластов, лейкоцитов крови, молекулярно-генетическим тестированием в сочетании с гистологическим исследованием ткани печени после биопсии. Прогноз для жизни для пациентов с этим заболеванием стал более обнадеживающим, когда появился препарат для ферментной заместительной терапии ДЛКЛ — себелипазы альфа.
- Болезнь накопления эфиров холестерина (БНЭХ). Проявляется в юношеском и взрослом возрасте, имеет более благоприятное течение. При БНЭХ поражается не только печень, но и сердечно-сосудистая и нервная система, надпочечники, другие органы, что определяет неспецифические клинические проявления и затрудняет диагноз во взрослом возрасте.
Жизнь с генетически обусловленными заболеваниями печени
Наличие генетической предрасположенности к развитию заболеваний печени может быть заподозрено в случае, если ближайшие родственники (обычно старшее поколение) также отмечают проблемы с печенью с раннего возраста: например, периодическое возникновение желтухи при отсутствии инфекционных заболеваний. Или если в семье были случаи ранних смертей от цирроза печени по неясным причинам.
Прогноз для жизни зависит от вида наследственно обусловленного нарушения обмена веществ в печени. В случаях наследственной гипербилирубинемии прогноз обычно благоприятный. При раннем выявлении болезней накопления меди и железа прогноз также может быть вполне благоприятным. Тем не менее, в некоторых случаях заболевание длительно протекает скрыто или нетипично, что затрудняет постановку диагноза вплоть до развития цирроза печени.
Доступность генетических методов тестирования позволяет с высокой точностью установить диагноз. В случае выявления наследственной патологии печени возможно тестирование ближайших родственников, а также детей пациента для выявления заболевания на ранних стадиях.
Вы можете задать вопрос врачу-гепатологу в комментариях. Спрашивайте, не стесняйтесь!
Статья обновлялась в последний раз: 11.06.2019
Не нашли то, что искали?
Автор-эксперт: Гастроэнтеролог-гепатолог Екатерина Кашух
Бесплатный путеводитель по знаниям
Подпишитесь на рассылку. Мы будем вам рассказывать, как пить и закусывать, чтобы не навредить здоровью. Лучшие советы от экспертов сайта, который читают больше 200 000 человек каждый месяц. Прекращайте портить здоровье и присоединяйтесь!
Источник
Болезнь Жильбера, она же – наследственный пигментный гепатоз, она же — доброкачественная гипербилирубинемия. Это заболевание печени, которое характеризуется наследственными нарушениями обмена билирубина с преобладанием в крови свободной фракции его, являющейся продуктом распада гемоглобина, эритроцитов крови.
Болезнь Жильбера — генетическое заболевание, передающееся по наследству, характеризуется изменением гена, участвующего в обмене билирубина. Повышение билирубина в крови развивается в связи с наследственной недостаточностью фермента глюкуронилтрансферазы, которая катализирует соединения билирубина с глюкуроновой кислотой в клетках печени.
Чаще болеют мужчины с 10 до 30 лет.
Болезнь Жильбера характеризуется периодическим умеренным повышением билирубина в анализе крови, появлением желтухи, чаще склер глаз.
Симптомы болезни Жильбера
Наблюдения показывают одновременное или одиночное проявление следующих симптомов:
- тупые боли в правом подреберье,
- тошнота, тяжесть под ложечкой,
- вздутие живота,
- непереносимость углеводов, алкоголя,
- слабость, недомогание,
- головные боли,
- раздражительность,
- нарушения сна (бессонница),
- трудности с концентрацией внимания.
При объективном обследовании редко наблюдается увеличение печени.
Диагностика болезни Жильбера
Из лабораторных показателей отмечается повышение билирубина и его фракций. Повышенный уровень билирубина более 40 мкмоль/л становится токсичным для печени и повышает уровень печеночных трансаминаз (АЛТ И АСТ), щелочной фосфотазы. В клиническом анализе крови нередко отмечается повышенное количество эритроцитов и гемоглобина, в анализе мочи определяется уробилин, а билирубина нет.
Важное значение в диагностике болезни Жильбера имеет молекулярная диагностика — генетический анализ ДНК в промоторной обл. UGT1.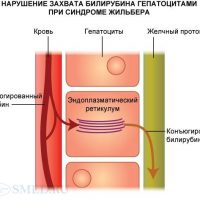
Ультразвуковое исследование печени и желчного пузыря не выявляет изменений, но как осложнения болезни может определяться желчнокаменная болезнь или признаки жировой печени различной степени.
При длительном течении болезни Жильбера показано проведение обследование эластометрии, выявляющее выраженность фиброза в печени.
Лечение болезни Жильбера
Лечение проводится амбулаторно в период обострения заболевания.
Лекарственные препараты – индукторы глюкуронилтрансферазы, которые уменьшают образование билирубина, нормализуют свободный (непрямой) билирубин: Зиксорин, Фенобарбитал 0.1 на ночь в течение месяца, Валокордин (Корвалол) по 20 капель на ночь 1 месяц с обязательным лабораторным контролем крови на билирубин.
Применение препаратов — гепатопротекторов (Карсил, Легалон, Урсосан), желчегонных препаратов (Хофитол, Лиф-52).
Лекарственные препараты назначаются врачом.
Питание осуществляется в объеме диеты 5, исключается алкоголь, курение, ограничиваются физические нагрузки, рекомендуется полупостельный режим, исключение лекарственных препаратов, обладающих гепатотоксическим действием (нестероидные противовоспалительные средства, циметидин, антибиотики, анальгетики, анаболические гормоны, гипотензивные средства, оральные контрацептивы и др.).
Болезнь Жильбера и жизнь с ней
Пациенты с болезнью Жильбера нуждаются в динамическом наблюдении: исследование крови на билирубин и печеночные пробы необходимо делать 2 раза в год, и при усилении иктеричности склер, и при обострении болезни. Профилактическое лечение 2 раза в год назначается индивидуально врачом и включает препараты, которые используются во время обострения.
Прогноз заболевания в целом благоприятный.
Источник