Фото людей у которых синдром tar

Анизокория
Анизокория — это состояние, при котором у человека наблюдается разница между размерами зрачков. При этом часто один зрачок имеет патологию, а именно, постоянно находится в зафиксированном положении.

Клинически важно установить, какой именно из зрачков находится в патологическом состоянии, так как это может указывать на наличие других отклонений. Например, отсутствие активности у меньшего из двух зрачков может быть признаком синдрома Горнера, который возникает при поражении нервной системы.
Гетерохромия
Еще одна аномалия, связанная с глазами — гетерохромия. Она выражается в различном цвете радужной оболочки правого и левого глаза или разной окраске определенных участков глаз. Возникает это нарушение от избытка или недостатка меланина — пигмента, который отвечает за цвет. У альбиносов он, к примеру, может вообще отсутствовать, а у людей с гетерохромией наблюдается в измененном состоянии.
Эта аномалия настолько популярна, что многие даже покупают разноцветные линзы, подражая людям с гетерохромией. А еще данная особенность очень привлекает представителей модельных агентств. Мы уже рассказывали о двух братьях из Турции, которые благодаря гетерохромии стали моделями.
Витилиго
О витилиго вы, наверное, уже наслышаны. Сейчас очень много моделей с этим заболеванием, и когда-то мы показывали вам фото самых популярных из них. Витилиго — это нарушение пигментации, которое выражается в исчезновении пигмента меланина на отдельных участках кожи.

Витилиго может начаться в любом возрасте в результате воздействия некоторых лекарственных и химических веществ, после воспалительных и некротических процессов на коже, из-за нервно-трофических, нейроэндокринных и аутоиммунных факторов. А вот предрасположенность к витилиго передается по наследству.
Синдром Шмида-Фраккаро
Синдром Шмида-Фраккаро, который также называют Синдромом кошачьего глаза — редкая врожденная патология, которая возникает из-за наличия дополнительной хромосомы. Один из характерных клинических признаков синдрома — верхняя коломба глаза, то есть отсутствие части радужной оболочки. Этот дефект делает взгляд человека похожим на кошачий, отсюда и название.
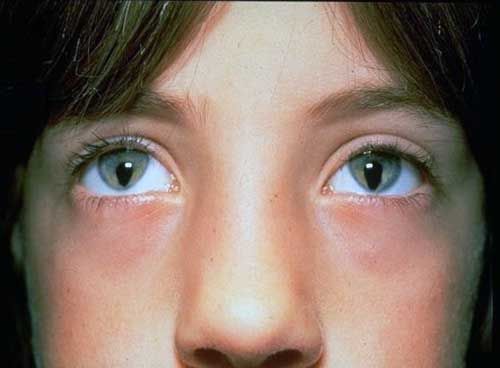
Особенность внешности — далеко не единственное, с чем приходится столкнуться больным синдромом Шмида-Фраккаро. У них могут наблюдаться пороки сердца, низкорослость, аномалии развития почек, умственная отсталость, сколиоз и многие другие проблемы.
Синдром нерасчесываемых волос
Синдром нерасчесываемых волос (еще одно его название — стекловидные волосы) делает его носителей похожими на одуванчики. Их шевелюра торчит во все стороны, при этом возможность уложить ее, собрать в пучок или заплести косу отсутствует в связи с огромным объемом, вызванным структурной аномалией.

Обычно заболевание развивается в грудном возрасте, а в период полового созревания проходит само. Но медицине известны случаи, когда этот синдром преследовал людей на протяжении всей жизни. И хоть сама по себе эта аномалия не представляет никакой опасности, согласитесь, приятного мало.
Смотрите также — «Дети не твои, и ты — не ты», или Как нерожденная женщина стала матерью четырех детей
Понравилось? Хотите быть в курсе обновлений? Подписывайтесь на наш Twitter, страницу в Facebook или канал в Telegram.
Источник
Источник
Синдром TAR
Эта статья для Медицинские специалисты
Профессиональные справочные статьи предназначены для использования медицинскими работниками. Они написаны британскими врачами и основаны на научных данных, британских и европейских руководствах. Вы можете найти один из наших статьи о здоровье полезнее.
Эта страница находится в архиве. Он не обновлялся с 20/04/2011. Внешние ссылки и ссылки могут больше не работать.
Синдром TAR
- эпидемиология
- презентация
- Дифференциальная диагностика
- исследования
- Сопутствующие заболевания
- управление
- осложнения
- Прогноз
- профилактика
Синонимы: T хромбоцитопения с bsent р синдром адиуса (следовательно, TAR), синдром тетрафомокелий-тромбоцитопении
эпидемиология
- Это редкое аутосомно-рецессивное состояние.
- Распространенность оценивается в 0,5-1 на 100 000 человек.1
презентация2
- Тромбоцитопения — может быть преходящим, симптомы обычно присутствуют на первой неделе жизни, с пурпурой, петехиями, носовым кровотечением, меланомой, кровохарканьем, гематурией, гематемезом.
- Голова и шея — внутричерепное кровотечение, затруднения в обучении, гипоплазия мозжечка, брахицефалия, микрогнатия, косоглазие, птоз, небольшой вздернутый нос, воспаление язвы на лбу.
- Нарушения верхних конечностей — двустороннее отсутствие радиусов, гипоплазия или отсутствие локтевой кости (может быть двусторонней), аномальная плечевая кость, гипоплазия запястной кости или слияние, гипопластические фаланги (но большие пальцы всегда присутствуют). Большие пальцы обычно имеют нормальный вид, но удерживаются в положении сгибания пястно-фалангового отдела.
- Живот — кисты поджелудочной железы, дивертикул Меккеля.
- Позвоночник и таз — расщелина позвоночника, задержка моторного развития, вывих бедра, тазобедренная кость.
- Аномалии нижних конечностей (47%) — подвывих коленного сустава, вывих надколенника, перекрут бедра или большеберцовой кости, малоберцовая кость может отсутствовать.
Дифференциальная диагностика1
- Синдром Фанкони
- Синдром Холта-Орама — аномалии верхних конечностей и сердца.
- Синдром RAPADILINO (относящийся к: лучевой гипоплазии / аплазии, гипоплазии надколенника / аплазии, расщелине или сильно выпуклому небу, диарее, вывихам суставов, небольшому росту и другим аномалиям).
- Синдром Робертса — пренатальное и постнатальное ограничение роста, черепно-лицевые аномалии, дефицит конечностей и гиперплазия половых органов.
- Трисомия 18 (синдром Эдвардса).
- Ассоциация VACTERL (= В пороки позвоночника, пороки развития носа, С сердечно-сосудистые дефекты, T рахео-пищеводные дефекты, (о)Е атрезия пищевода, р анальные отклонения, L imb (уродства)) — синдром врожденных аномалий, включая дисгенез позвонков и аномалии почек и конечностей.
исследования
Лабораторные тесты3
- FBC может показать:
- Низкое количество тромбоцитов — 15-30 х 109/ Л.
- Эозинофилия (у 50% пациентов).
- Лейкоцитоз — WBC больше 35 х 109/ Л с левым смещением, лейкоэмоидная реакция.
- Анемия вторичная по отношению к кровотечению.
- Генетические тесты — хотя считается, что это аутосомно-рецессивное состояние, генетическое тестирование обычно нормальное, что помогает дифференцировать его от других врожденных состояний (см. Дифференциальный диагноз выше)
Радио-изображения
Ультразвук — трансвагинальное ультразвуковое исследование может выявить вовлечение скелета уже в 13 недель беременности. Наличие радиальной аплазии свидетельствует о необходимости сканирования аномалий лица, конечностей и почек, чтобы дифференцировать это состояние от других синдромов.4
процедуры
- Кордоцентез для отбора образцов плода может обнаружить тромбоцитопению пренатально.4 Риски:
- 1-2% потеря плода.
- Длительное кровотечение из места прокола.
- Отбор проб костного мозга может выявить:
- Нормальный или гиперклеточный костный мозг.
- Уменьшенные, отсутствующие или незрелые мегакариоциты.
- Мелкие, базофильные, вакуолизированные мегакариоциты.
- Эритроидная гиперплазия.
Сопутствующие заболевания 2, 5
- Непереносимость коровьего молока.
- Почечные нарушения, например, подковообразная почка.
- Сердечные нарушения, например, дефект межпредсердной перегородки (ASD), дефект межжелудочковой перегородки (VSD), тетралогия Фалло.
управление
медицинская
- Пренатальное переливание тромбоцитов было успешно выполнено у плода, у которого была выявлена тяжелая тромбоцитопения при кордоцентезе.6
- Общие меры по снижению риска кровотечения должны быть приняты, если количество тромбоцитов падает ниже 80 х 109/ Л.3 Они включают:
- Предотвращение травм, например, дети должны носить мягкий шлем.
- Избегание антиагрегантных препаратов, эгаспирина и нестероидных противовоспалительных препаратов (НПВП).
- Длительное давление на места инъекции.
- Острое кровотечение из-за травмы конечности должно лечиться давлением и подъемом конечности. Пациент должен быть в тепле и доставлен в больницу как можно скорее.
- Переливание тромбоцитов следует учитывать, если риск переливания крови (инфекция, анафилаксия, гемолитическая реакция) заменяется риском значительного кровотечения.3 Руководящий принцип (взятый из управления тромбоцитопенией) — количество тромбоцитов ниже 40 х 109).
- Сниженные лейкоцитами концентрации тромбоцитов могут быть указаны у пациентов с риском тяжелого кровотечения.
- Пациентам, которые не отвечают на переливание, могут помочь HLA-подобранные тромбоциты от членов семьи, хотя это не гарантирует успеха, поскольку могут происходить антигенные реакции не-HLA.
- Трансплантация костного мозга должна рассматриваться для пациентов, устойчивых к переливаниям тромбоцитов.
Другие меры
- Физиотерапия и трудотерапия могут помочь улучшить функцию.
- Хирургическая коррекция деформаций может быть необходимым. Этому иногда предшествует шинирование пораженной части для улучшения функции.
- Если операция невозможна, цель должна состоять в том, чтобы как можно лучше улучшить независимость и функционирование. Могут потребоваться адаптивные устройства и вспомогательные средства для стирки, одевания и кормления.
- Протезы, как правило, не помогают из-за мышечной слабости.
- Спленэктомия обычно является эффективным лекарством от тромбоцитопении, сохраняющейся во взрослой жизни.
осложнения
Осложнения обычно связаны с кровотечением, особенно внутричерепное кровоизлияние.
Прогноз
- Если пациенты выживают в первые два года жизни, продолжительность жизни нормальная.
- Основной причиной смертности является кровотечение.
- Кровотечение, особенно внутричерепное кровоизлияние, может привести к значительной заболеваемости.
- Прогноз в отношении функции кисти и верхней конечности обычно хороший, при условии, что радиальная аплазия является единственной аномалией скелета.
профилактика
Генетическое консультирование и ранняя пренатальная диагностика повышает вероятность прерывания, если будет показано, что плод поражен.7 Методика, называемая сравнительной геномной гибридизацией, — метод оценки ошибок в воспроизведении ДНК, связанных с этим состоянием, — значительно помогла процессу генетического консультирования.8
Считаете ли вы эту информацию полезной? да нет
Спасибо, мы только что отправили электронное письмо с опросом, чтобы подтвердить ваши предпочтения.
Дальнейшее чтение и ссылки
Toriello HV; Синдром отсутствия радиуса тромбоцитопении
Синдром радиуса без тромбоцитопении, он-лайн менделевское наследование у человека (OMIM)
Ву Дж. К. и др., Синдром отсутствия радиуса при тромбоцитопении, Medscape, сентябрь 2009 г.
Донненфельд А.Е., Вайзман Б., Лави Е. и др.; Пренатальная диагностика синдрома отсутствия радиуса тромбоцитопении с помощью ультразвука и кордоцентеза. Пренат Диаг. 1990 янв. 10 (1): 29-35.
Гринхалх К.Л., Хауэлл Р.Т., Боттани А. и др.; Синдром радиуса отсутствия тромбоцитопении: клиническое генетическое исследование. J Med Genet. 2002 Dec39 (12): 876-81.
Вайнблатт М., Петриковский Б., Биалер М. и др.; Пренатальная оценка и переливание тромбоцитов при тромбоцитопении при отсутствии радиуса синдрома. Пренат Диаг. 1994 сент. 14 (9): 892-6.
Ward RE, Bixler D, Provisor AJ и др.; Передача от ребёнка синдрома отсутствия радиуса тромбоцитопении (TAR). Am J Med Genet Suppl. 19862: 207-14.
Uhrig S, Schlembach D, Waldispuehl-Geigl J и др.; Влияние массива сравнительной информации, полученной из геномной гибридизации, на генетическое консультирование, продемонстрированное пренатальной диагностикой микроделеции, связанной с синдромом TAR (тромбоцитопения-отсутствующий радиус), 1q21.1. Am J Hum Genet. 2007 Октябрь (4): 866-8.
Следующая статья
Hyperaldosteronism
Источник
 Срез костного мозга, на котором чёрными стрелками отмечены мегакариоциты – клетки, продуцирующие тромбоциты. (кликните картинку для увеличения) |
Учёные наконец-то определили генетические предпосылки, ответственные за тромбоцитопению с отсутствием лучевой кости (TAR-синдром или ТАР-синдром) – редкое генетическое заболевание, при котором у пациента отмечается тромбоцитопения и отсутствие лучевой кости. В настоящее время специалисты пытаются внедрить результаты проведённого исследования в практическую деятельность.
В ходе проведения исследования специалисты смогли установить, что TAR-синдром является следствием низкого содержания белка Y14. Так же они выяснили, что болезнь развивается с помощью уникального наследуемого механизма.
Тромбоциты содержаться в нашем организме в очень большом количестве. Они занимаются поиском повреждений стенок сосудов и устраняют их. У некоторых людей от рождения в организме содержится ненормально малое количество тромбоцитов. Предполагается, что ненормально низкое содержание тромбоцитов передаётся по наследству. ТАР-синдром объединяет в себе уникальные характерные черты низкого уровня содержания тромбоцитов, значительные кровотечения (особенно в раннем детстве) и скелетных аномалий, затрагивающих верхние конечности (от отсутствия радиальной кости в предплечье до практически полного отсутствия верхних конечностей). На протяжении 50 лет учёные пытались установить генетические основы развития данного заболевания.
«Без использования современных технологий для проведения генетических исследований было бы гораздо сложнее установить особенности наследования болезни» — говорит доктор Корнелис Альберс (Cornelis Albers, институт Сенгера, Кембриджский университет). «Для того, чтобы получить определённый результат мы расшифровали 40 миллионов знаков генетического кода у 5 пациентов» — продолжает он.
Многие люди, у которых отмечается наличие TAR-синдрома, несут делецию в одной из копий 1-й хромосомы. Однако, как было принято считать, данная делеция не является основной причиной развития болезни (дело в том, что у родительских организмов, несущих эту мутацию, не отмечается развитие TAR-синдрома). Следовательно, должна быть иная причина. Учёные расшифровали геном людей, у которых отмечается наличие ТАР-синдрома и в геноме присутствует названная выше делеция. Специалисты выяснили, что подавляющее большинство испытуемых характеризовалось наличием одной из двух мутантных копий гена под названием RBM8A. Они установили, что когда ребёнок одновременно наследует и делецию в одной из копий хромосомы 1, и одну копию мутантного гена RBM8A, то у него отмечается развитие TAR-синдрома.
RBM8A контролирует продукцию белка Y14. Учёные обнаружили, что при комбинации мутаций (делеции одной из копий RBM8A и изменении другой копии) происходит значительное падение уровня содержания Y14. Авторы исследования пришли к выводу, что резкое падение концентрации Y14 является причиной развития TAR-синдрома, влияя на образование тромбоцитов.
«Невозможность продукции Y14 в необходимом количестве у пациентов, у которых наблюдается TAR-синдром, влияет на формирование тромбоцитов, но не затрагивает формирование других клеток крови» — говорит доктор Седрик Гиверт (Cedric Ghevaert, учёный из Кембриджского университета). «Нам в некоторой степени удалось показать то, каким образом некоторые передающиеся по наследству болезни, могут проявляться в столь явных чертах, имеющих отношение к, на первый взгляд, несвязанным между собой событиям: дефектам развития костей скелета и дефектам функционирования крови» — уточняет он.
Александр Шустер
Также по теме:
- Ген insomniac у дрозофил оказывает влияние на способность спать
- Получены данные о влиянии гена SIRT1 на развитие болезни Хантингтона
Источники:
- Новость подготовлена по материалам ресурса www.sciencedaily.com
самое популярное
- пишут
- читают
- оценивают
Лечение наркомании частный наркологический центр лечения.
Источник
Синдром TAR у плода. Синдром Aase, Холта-Орама плода
Синдром TAR (тромбоцитопении — аплазии лучевой кости) является аутосомно-рецессивным заболеванием, которое характеризуется тромбоцитопенией (количество тромбоцитов меньше 100 000/мм3) и двусторонней аплазией лучевой кости. Большие пальцы и пястные кости кистей при этом состоянии всегда присутствуют; могут быть редуцированы локтевая и плечевая кости и всегда обнаруживается лучевая форма косорукости.
Пороки сердца выявляются в 33% случаев (например, тетрада Фалло (Fallot) и дефекты перегородок). В подобных ситуациях рекомендуется проводить родоразрешение путем операции кесарева сечения из-за риска возникновения внутричерепного кровоизлияния у плода. Описано большое количество наблюдений пренатальной диагностики синдрома тромбоцитопении — аплазии лучевой кости.
Синдром Aase является аутосомно-рецессивным заболеванием и характеризуется врожденной гипопластической анемией, лучевой косорукостью с двухсторонними трехфаланговыми большими пальцами кистей и гипоплазией дистальных отделов лучевых костей. Также могут наблюдаться дефекты межжелудочковой перегородки и коарктация аорты.
Трехфаланговые большие пальцы являются характерным признаком нескольких видов костных дизостозов и синдромов аномалий развития. В виде изолированных пороков они также могут находиться в случайной ассоциации с другими аномалиями и часто характеризуются семейным типом наследования. К другим заболеваниям, при которых могут выявляться трехфаланговые большие пальцы кистей, относятся синдром Holt-Oram, синдром Diamond-Blackfan, хромосомные аномалии и гидантоиновый синдром плода.
Синдром Холта-Орама (Holt-Oram) представляет собой аутосомно-доминантное заболевание, которое характеризуется врожденными пороками сердца (главным образом, дефектами межпредсердной перерегородки вторичного типа и дефектами межжелудочковой перегородки), аплазией или гипоплазией лучевой кости и трехфаланговыми большими пальцами кистей или их аплазией.
Дефекты конечностей часто бывают асимметричными, причем преимущественно поражается левая сторона. Не было выявлено корреляции между выраженностью дефектов конечности и наличием аномалий сердца. Действительно, некоторые пациенты с этим синдромом имеют только скелетные аномалии. Также могут обнаруживаться гипертелоризм и пороки развития грудной клетки и позвонков.
Описана пренатальная диагностика этого заболевания. Мутация, которая вызывает синдром Holt-Oram, недавно была идентифицирована. Синдром, проявляющийся сочетанием пороков верхних конечностей и сердечно-сосудистой системы, описанный К.В. Lewis et al., возможно, является разновидностью синдрома Holt-Oram.
— Также рекомендуем «Ассоциация VATER у плода. Синдром Голденхара и полидактилия плода»
Оглавление темы «Пороки развития скелета плода»:
1. Кампомелическая дисплазия у плода. Диагностика кампомелической дисплазии
2. Гипоплазия грудной клетки плода. Хондроэктодермальная дисплазия
3. Врожденные ампутации у плода. Синдром аглоссии-адактилии
4. Редукционные пороки конечностей у плода. Фокомелия у плода
5. Врожденное укорочение бедренной кости. Расщепление кистей и стоп плода
6. Аномальная установка кистей плода. Анемия Фанкони у плода
7. Синдром TAR у плода. Синдром Aase, Холта-Орама плода
8. Ассоциация VATER у плода. Синдром Голденхара и полидактилия плода
9. Артрогрипоз у плода. Распространенность артрогрипоза у плода
10. Диагностика артрогрипоза плода. Фетальные синдромы
Источник