Фото лица с синдромом марфана

В последние годы отмечается значительный рост наследственных заболеваний с поражением соединительной ткани. Некоторые специалисты считают, что причина такого явления кроется в накоплении в процессе эволюции человека новых генетических изменений (мутаций), к чему приводят внутренние и внешние факторы окружающей среды. Другие полагают, что на самом деле просто возросли диагностические возможности в связи с совершенствованием медико-генетических знаний, которые позволяют распознать генетическую патологию и поставить правильный диагноз.
Большую группу наследственных заболеваний составляет поражение соединительной ткани. Так как она является неотъемлемой частью всех органов и систем в организме, то такие патологии отличаются множеством нарушений и клинических симптомов. Одним из самых известных генетических заболеваний с поражением соединительной ткани считается синдром Марфана.
Такая патология характеризируется большой вариабельностью симптоматики (от скрытых форм до несовместимых с жизнью вариантов течения) и чаще всего включает поражения сердечно-сосудистой системы, глаз, центральной нервной системы и опорно-двигательного аппарата.
Встречается заболевание достаточно редко – 1 случай на 10 000 человек. Но если обратиться к статистике европейских стран, то частота значительно выше – 1-3 случая на 5000 человек, что связано с большей доступностью специфической диагностики скрытых форм патологии.

Известные люди тоже болели синдромом Марфана
Причины и генетика патологии
Впервые болезнь детально описал в 1896 году французский педиатр А.Марфан, в честь которого и назвали патологию. Он наблюдал за 5-летней девочкой астенического телосложения с непропорционально длинными конечностями и врожденной арахнодактилией (длинными пальцами). К середине 20-х годов прошлого столетия уже имелось множество подобных описанных клинических случаев у детей и взрослых. Американский генетик Мак Кьюсик провел детальное исследование мутаций хромосом и открыл новую группу наследственных заболеваний соединительной ткани, куда и отнесли синдром Марфана.
По этиологии синдром Марфана является генетическим заболеванием с аутосомно-доминантным типом наследования, с различной степенью экспрессивности (клинических проявлений генетических изменений). Примерно 85% случаев недуга носит наследственный характер, то есть наследуется от родителей. Остальные случаи являются новыми, то есть возникают вследствие новых спонтанных мутаций, а не передаются по наследству.
Непосредственная причина патологии в 95% случаев – это мутация в гене, которые кодирует строение фибриллина-1 и/или фибриллина-2. Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.

Синдром Марфана наследуется по аутосомно-доминантному типу
Фибриллин – это основа эластических волокон соединительной ткани гликопротеиновой природы. Он составляет каркас межклеточного вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других органов и тканей. В случае наличия описанной мутации у пациента соединительная ткань отличается повышенной способностью к растяжению, становится менее прочной и выносливой к механическим воздействиям, что и становится причиной клинических проявлений синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана (атипичные формы патологии) является точковая мутация гена, который кодирует строение α2-цепи коллагена первого типа.
Классификация
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.

Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.
Симптомы синдрома Марфана
Признаки синдрома Марфана очень разнообразны. Поэтому их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- органов зрения;
- сердечно-сосудистой системы;
- нервной системы;
- дыхательной системы;
- кожных покровов и мягких тканей;
- прочих органов и систем.
Опорно-двигательный аппарат
Патологическая соединительная ткань обусловливает развитие ряда специфических фенотипических признаков и деформаций скелета у пациентов с данной патологией.

Характерный внешний вид пациентов с синдромом Марфана
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически. Для них характерно:
- астеническое телосложение;
- высокий рост;
- плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
- очень длинные верхние и нижние конечности при относительно коротком туловище;
- череп вытянутый (долихоцефалический);
- удлиненные пальцы – паукообразные (арахнодактилия);
- лицо узкое, вытянутое по вертикали;
- готическое верхнее небо;
- недоразвитие скул;
- выступающая нижняя челюсть (прогнатизм);
- неправильный рост (скученность) зубов и патологический прикус;
- гипермобильность суставов, их «разболтанность;
- глубоко посажены в черепе глаза.

Пример того, как определить наличие арахнодактилии, характерной для синдрома Марфана
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический кифоз и лордоз, осанку по типу «прямой спины» (сглаживание физиологического поясничного лордоза). Также появляются подвывихи и вывихи в шейном отделе позвоночника, примерно у 20% больных диагностируют поясничный спондилолистез.

Выраженная степень арахнодактилии при синдроме Марфана со специфическими контрактурами пальцев
Среди других скелетных деформаций встречается:
- протрузия вертлужной впадины тазобедренного сустава 2-3 степени, которая становится причиной диспластического коксартроза и инвалидности многих пациентов, если не выполнить операцию эндопротезирования;
- килевидная и вдавленная деформация грудной клетки;
- плоскостопие (продольное и поперечное).
Для пациентов в СМ также характерна остеопения (снижение минеральной плотности костей) и частые патологические переломы костей на ее фоне, а также склонность к привычным вывихам, например, плеча.
Вдавленная деформация грудной клетки у пациента с синдромом Марфана и результат ее хирургической коррекции
Сердечно-сосудистая система
Среди поражений кардиоваскулярной системы при СМ чаще всего встречаются:
- пролапс створок митрального клапана с регургитацией или без
- миксаматоз сердца;
- дилятационная кардиомиопатия с развитием сердечной недостаточности;
- аневризмы аорты и других сосудов (мозговых, почечных, пр.);
- расширение легочной артерии и различных отделов аорты.
Именно кардиоваскулярные патологические изменения при СМ определяют прогноз и продолжительность жизни пациентов. Примерно 90% всех пациентов с данной генетической патологией умирают в возрасте 40-50 лет вследствие таких осложнений, как расслоение и разрыв аневризмы аорты, других сосудов, прогрессирующей недостаточности сердца вследствие дилатации его камер и изменений клапанного аппарата.
При наличии СМ возможно наличие врожденных пороков сердца у детей. Чаще всего встречаются коарктация аорты, стеноз (сужение) легочной артерии, дефект межжелудочковой и межпредсердной перегородки.
Также такие пациенты склонны к различным сердечным аритмиям, среди которых и опасные для жизни (мерцательная аритмия, желудочковая тахикардия и экстрасистолия), к инфекционному эндокардиту.

Аневризмы аорты и их разрыв чаще всего становятся причиной смерти пациента с синдромом Марфана
Орган зрения
Патологические изменения глаз являются весьма характерными для данного недуга. Примерно у 60-80% пациентов диагностируется дислокация хрусталика из-за слабости его связочного аппарата, причем еще в младенческом возрасте. Среди других характерных признаков:
- уплощение роговицы;
- увеличение размеров глазного яблока в длину;
- миопия или гиперметропия;
- нарушение процесса аккомодации из-за недоразвития цилиарной мышцы.
В случае выявления описанных поражений органа зрения у новорожденного ребенка следует задуматься о возможной генетической патологии.

Дислокация хрусталика – типичный признак синдрома Марфана
Нервная система
Из-за патологического строения стенок сосудов у пациентов с СМ повышен риск геморрагических инсультов, а также кровоизлияний в мозг при разрыве сосудистых аневризм, субарахноидальных кровотечений.
Среди аномалий развития встречается эктазия твердой мозговой оболочки. Чаще всего приходится сталкиваться с пояснично-крестцовой эктазией мозговой оболочки (выпячивание твердой мозговой оболочки за пределы позвоночного канала через дефект в строении позвонков). Это большой критерий СМ, который встречается в 40% случаев заболевания.
У части пациентов встречаются отклонения в интеллектуальном развитии, но большинство людей с СМ характеризируются высокими показателями IQ.
Органы системы дыхания
В большинстве случаев изменения бронхолегочного аппарата диагностируются случайно. Характерно развитие булл в верхних частях легких, которые иногда могут разрываться с развитием спонтанного пневмоторакса.
Также из-за деформаций грудной клетки пациенты склонны к развитию эмфиземы легких, частых инфекционных заболеваний органов дыхания и дыхательной недостаточности.

Буллезная эмфизема может быть причиной спонтанного пневмоторакса у пациентов с синдромом Марфана
Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова, которая сочетается с развитием атрофических стрий. Последние появляются спонтанно, они никак не связаны с колебанием веса, беременностью или гормональными нарушениями. Подкожный жир выражен слабо у пациентов с синдромом Марфана. Они часто страдают рецидивирующими грыжами передней брюшной стенки.
Встречаются и многие другие патологические симптомы поражения прочих органов и тканей при СМ. Например, опущение почек (нефроптоз), выпадение мочевого пузыря и матки у женщин, варикозное расширение вен, хронические запоры и др.

Диагностика синдрома Марфана
Диагностика при синдроме Марфана носит в основном клинический характер. Обязательно учитывают анамнез, в том числе и семейный (наличие подобных проблем у кого-то из родственников), данные объективного обследования и осмотра. Также проводят множество дополнительных диагностических процедур для выявления патологии тех или иных органов и систем. Для этого применяют ЭКГ, УЗИ сердца и сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних органов, позвоночника, головного мозга, офтальмоскопию и прочие исследования органа зрения, аортографию, ангиографию и много других методик, в зависимости от клинической ситуации и симптомов болезни.
Существуют общепринятые диагностические критерии синдрома Марфана (большие и малые), которые позволяют с большой степенью вероятности поставить правильный, но предварительный диагноз пациенту.

Окончательный диагноз синдрома Марфана выставляют только после анализа генотипа (ДНК-диагностика) и выявления специфической мутации в гене, ответственном за продукцию фибриллина, с помощью молекулярно-генетических методик.
Лечение заболевания
К сожалению, на сегодняшний день вылечить синдром Марфана, как и повлиять на его причину, невозможно. Терапия в основном направлена на улучшение качества жизни больного человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и может включать как консервативные, так и хирургические методики.
Всем пациентам с СМ рекомендуют ограничения в физической активности к среднему или низкому уровню, избегать тяжелого физического труда, не заниматься спортом, так как это способствует прогрессированию патологии и травматизму.
Больные должны наблюдаться у различных докторов: кардиолога, окулиста, травматолога-ортопеда, клинического генетика, невролога и др.

Лечением синдрома Марфана должна заниматься целая команда специалистов
В случае выявления тех или иных кардиоваскулярных проблем назначают комплексное лечение, медикаменты для профилактики осложнений. Проводят медикаментозную коррекцию аритмий, частоты сердечных сокращений, артериального давления. Обязательно всем пациентам назначают прием бета-блокаторов, если нет противопоказаний к этой группе средств, которые имеют протекторный эффект в отношении возможного расслоения аорты.
В случае патологии клапанного аппарата сердца, аневризме аорты и других сосудов, расслоении их стенок может понадобиться операция. Также хирургическое вмешательство используют для коррекции деформаций опорно-двигательного аппарата.
Рекомендуемые специалистами схемы терапии больных с СМ в обязательном порядке включают коллаген-образующие и метаболические средства, препараты для восполнения макро- и микроэлементов.
В случае проблем с органом зрения также проводят оперативные вмешательства при эктопии хрусталика, лазерную коррекцию зрения, подбирают очки или контактные линзы.
В целом подбор лечебных методик и спектр применяемых средств очень индивидуальны. Это полностью зависит от присутствующих симптомов и степени выраженности болезни у конкретного пациента.

Основная задача в лечении пациентов с Синдромом Марфана заключается в предотвращении сердечно-сосудистых повреждений
Прогноз
Синдром Марфана отличается, как правило, хроническим прогрессирующим течением. Продолжительность жизни пациентов при условии полноценного комплекса лечебных мероприятий в среднем составляет 45 лет. Основными факторами риска преждевременной смерти выступают осложнения, которые возникают вследствие патологии кардиоваскулярной системы.
Синдром Марфана и беременность
Пациентки с СМ могут иметь детей, причем здоровых, но это очень опасно по двум причинам:
- Беременная женщина имеет очень высокий риск летальных осложнений СМ со стороны сердечно-сосудистой системы. Так как при вынашивании ребенка создается усиленная нагрузка на организм матери, а особенно на сердце и сосуды, то очень большие шансы у пациенток с синдромом Марфана получить разрыв аневризмы, ее формирование, расслоение аорты и прочие смертельно опасные осложнения.
- Риск передачи данной наследственной патологии своему ребенку составляет 50%.
Поэтому специалисты не рекомендуют беременеть женщинам с синдромом Марфана.
Возможна ли профилактика болезни?
К сожалению, специфической профилактики синдрома Марфана не существует. Семейная пара, в которой один или оба родителя страдают данной патологией, в обязательном порядке должны планировать беременность и пройти медико-генетическое консультирование у врача-генетика. Вместе с этим можно проводить пренатальную (дородовую) диагностику на предмет наличия СМ у плода.
Источник
Синдром Марфана — это генетическое заболевание, при котором патологические изменения затрагивают соединительную ткань. Из-за этого у пациента развиваются нарушения в строении скелета и работе сердца, а также ухудшается зрение. Такая болезнь встречается одинаково часто и у мужчин, и у женщин. Патология отмечается в 1 случае на 10000 новорожденных. В дальнейшем болезненные изменения прогрессируют и без лечения существенно сокращают продолжительность жизни пациента.
Причины заболевания
Основной причиной синдрома Марфана являются врожденные изменения в гене FBN1. Он контролирует выработку в организме белка фибриллина. Это вещество делает соединительную ткань эластичной и прочной.
В организме больных этот белок продуцируется в очень малом количестве или совсем не вырабатывается. Его дефицит негативно сказывается на состоянии соединительной ткани. Она слишком сильно растягивается, становится слабой и плохо справляется даже с небольшими физическими нагрузками. Из-за этого у людей с синдромом Марфана неправильно развивается скелет, отмечаются серьезные нарушения в работе сердечных клапанов. Вследствие подвижности хрусталика глаза наблюдается ухудшение зрения. Дефекты в соединительной ткани происходят еще во время внутриутробного периода. В течение жизни они прогрессируют. Без терапии это заболевание может привести к ранней смерти пациента.
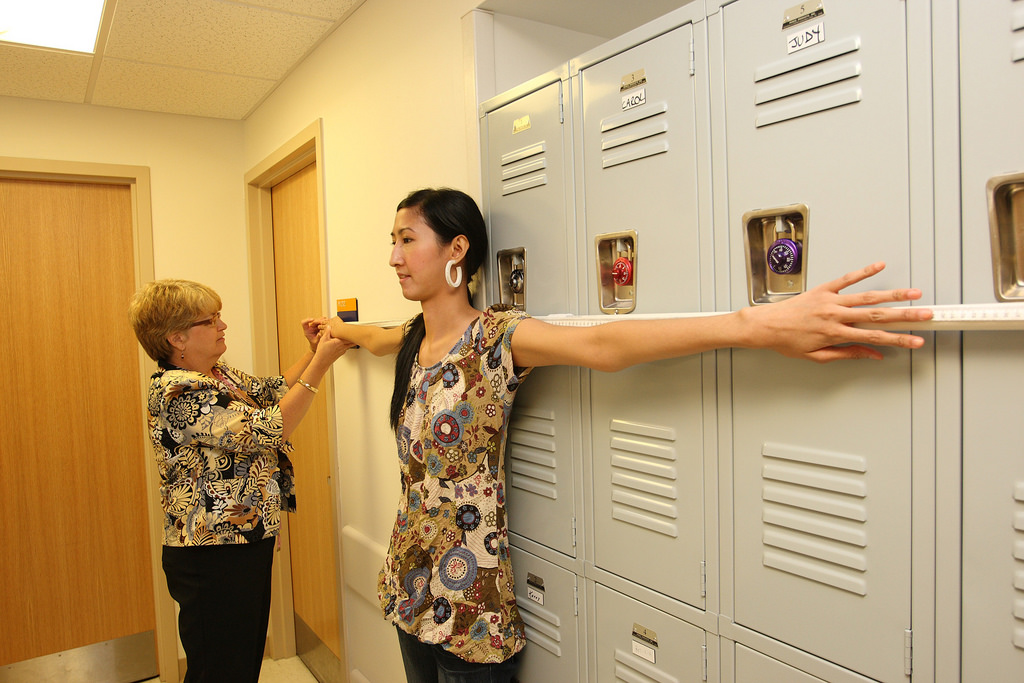
Как наследуется патология?
Наследование синдрома Марфана происходит по аутосомно-доминантному типу. Если болен один родитель, то он может передать патологию ребенку в 50% случаев. Если же этим синдромом страдают и отец, и мать, то вероятность рождения больного малыша увеличивается до 75-100%.
Чаще всего патология передается от больных родителей. Но бывает и так, что отец и мать здоровы, однако ребенок страдает синдромом Марфана. Это отмечается в 15% случаев. Причины такой случайной мутации гена неизвестны. Однако специалистами установлено, что риск рождения ребенка с такой патологией увеличивается, если возраст отца превышает 35-40 лет. Возраст матери в этом случае не влияет на мутацию гена.
Признаки поражения опорно-двигательного аппарата
Изменения в скелете являются одним из ведущих симптомов синдрома Марфана. Опытный врач может предположить наличие этой патологии уже во время осмотра по внешнему виду больного.
Люди с этим заболеванием обычно имеют высокий рост. У них тонкие кости с повышенной гибкостью в суставах и сухожилиях. При большом росте пациенты обычно имеют низкий вес и выглядят высокими и худощавыми. Кости удлиняются из-за чрезмерного растяжения соединительной ткани при синдроме Марфана. Фото больного можно увидеть ниже.
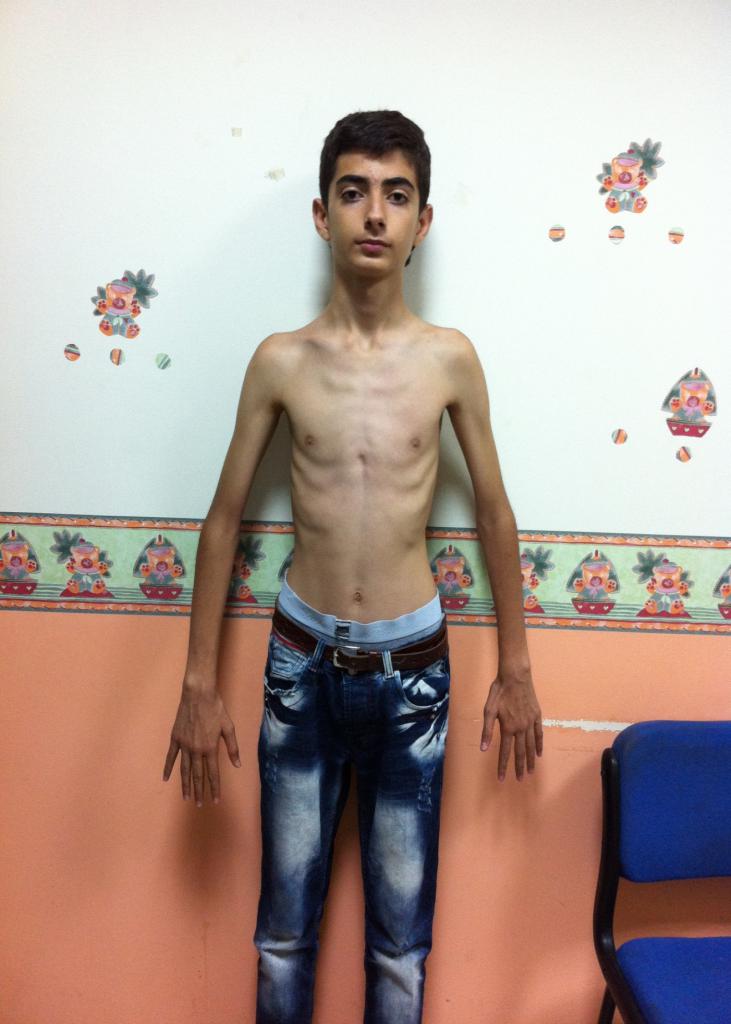
Конечности больных непропорционально длинные и тонкие. Некоторые пациенты, имея высокий рост и большой размах рук, пытаются заниматься спортом. Однако при такой патологии физические нагрузки категорически противопоказаны. Соединительная ткань из-за недостатка фибриллина становится очень слабой и может не выдержать напряжения. При высоком росте больные не отличаются большой физической силой, напротив, их кости и мышцы плохо справляются с нагрузкой.
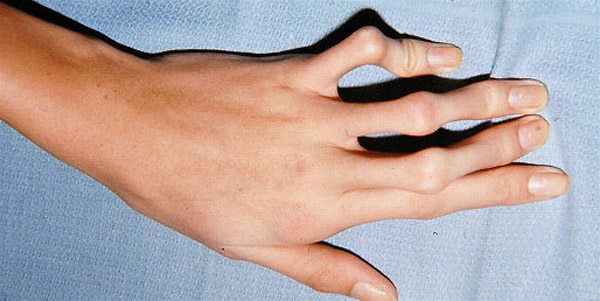
Другим признаком патологии являются непропорционально длинные пальцы. Они обладают повышенной гибкостью. Такой симптом называют «паучьими пальцами», или арахнодактилией, это одно из характерных внешних проявлений синдрома Марфана. Фото кисти больного можно увидеть ниже.
У страдающих этим синдромом лицо имеет длинную и узкую форму. Есть изменения в небе, оно высокое и изогнутое. Из-за этого у больного ребенка могут неправильно расти зубы. У взрослых меняется голос, появляется хрипота.
Жировая ткань у пациентов мало развита, из-за этого они имеют пониженную массу тела и выглядят очень худыми при высоком росте. Мышцы слабые, плохо выдерживают нагрузку.
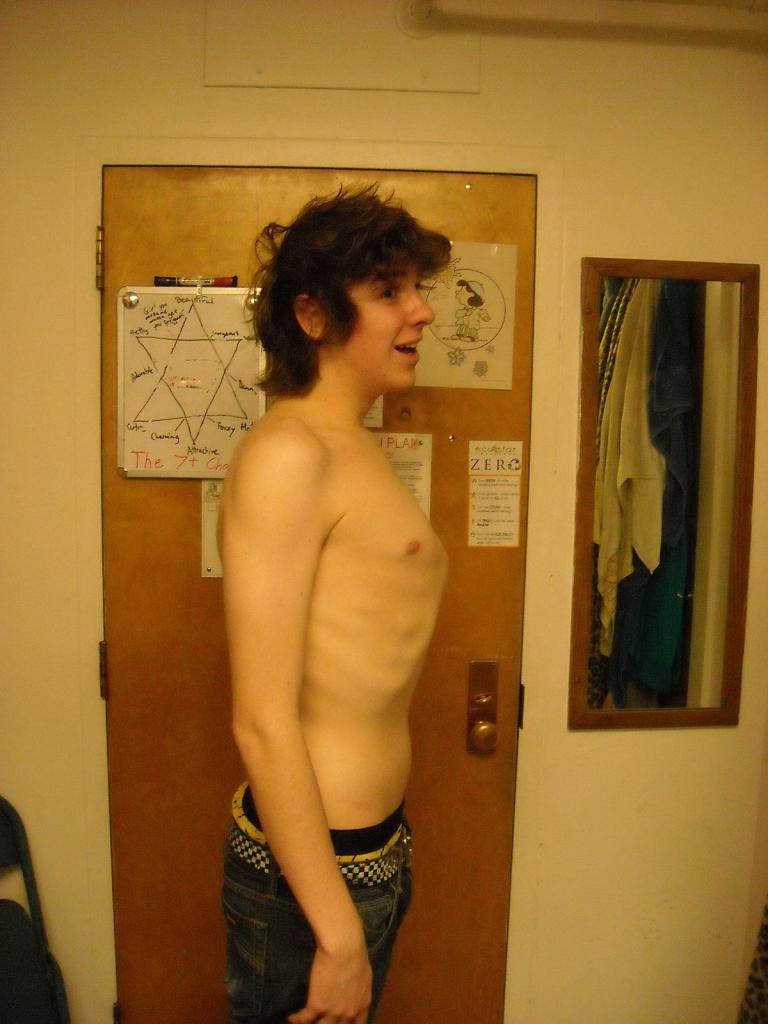
У больных отмечается плоскостопие, искривления позвоночника и деформации костей туловища. Вдавленная или выступающая грудная клетка — частое проявление синдрома Марфана. На фото можно увидеть деформацию костей у больного.
Ухудшение зрения
Из-за большой растяжимости соединительной ткани повышается подвижность хрусталика глаза. Нередко происходит вывих или подвывих этого органа. При осмотре у окулиста определяется сильное смещение хрусталика. Это негативно сказывается на зрении. У пациентов с синдромом Марфана выявляются следующие офтальмологические нарушения:
- близорукость;
- глаукома;
- катаракта.
Эти заболевания возникают еще в детстве. Часто ребенку с малых лет приходится носить очки. Опасным осложнением смещения хрусталика может стать отслойка сетчатки, которая приводит к сильному ухудшению зрения.

Симптомы со стороны сердца
Поражение сердца является наиболее опасным признаком синдрома Марфана. Кардиологические патологии нередко становятся причиной летального исхода из-за разрыва аорты. У пациента отмечаются следующие болезненные изменения:
- расширение и аневризма аорты;
- нарушение работы сердечных клапанов;
- увеличение сердца;
- нарушение проводимости миокарда.
Из-за этих отклонений наблюдаются боли в груди по типу стенокардии, одышка, аритмия, тахикардия, ощущение постоянной усталости. Иногда дети, унаследовавшие этот синдром, рождаются с пороками сердца, что приводит к гибели на первом году жизни.
Поражение нервной системы
При синдроме Марфана симптомы со стороны центральной нервной системы связаны с набуханием мешочка (оболочки), который окружает спинной мозг. Это образование также состоит из соединительной ткани, которая патологически изменена. Врачи-невропатологи называют такое нарушение дуральной эктазией. Сначала человек испытывает лишь небольшой дискомфорт. Но со временем патология прогрессирует, и усиливается давление оболочки на нижние отделы спинного мозга. Больного беспокоят частые боли в животе, а также слабость и онемение ног.
Изменения в легких
Патологические изменения в легких отмечаются не всегда. Однако эластичность соединительной ткани нарушена и в органах дыхания. В результате этого повышается риск возникновения пневмоторакса, дыхательной недостаточности и эмфиземы легких. У больных нередко происходит спонтанная остановка дыхания (апноэ) в ночное время.
Другие проявления
У пациента появляются растяжки на коже, которые никак не связаны с изменением массы тела. Это является лишь косметическим недостатком и обычно не доставляет никакого беспокойства.
Однако пациенты очень склонны к образованию грыж в паховой и брюшной области, которые часто рецидивируют, несмотря на лечение.
Этот синдром нередко сопровождается эндокринными нарушениями, у больных повышен риск возникновения сахарного диабета и акромегалии. Из-за растяжения соединительной ткани происходит опущение мочеполовых органов. Пациенты часто получают травмы: вывихи и растяжения сухожилий.
Общее самочувствие
При этой генетической патологии больные чувствуют себя постоянно уставшими. Они утомляются даже от незначительного физического напряжения. Мускулатура их слабо развита, после нагрузки пациенты ощущают боли в мышцах. Такие признаки связаны с поражением соединительной ткани.
При психоэмоциональном напряжении могут возникать головные боли по типу мигрени, астения и понижение АД.
Страдает ли умственное развитие
У больных детей не отмечается задержки умственного развития. Пациенты с синдромом Марфана имеют нормальный интеллект. В некоторых случаях их уровень IQ даже выше среднего. Однако у больных иногда наблюдаются признаки повышенной нервозности: излишняя эмоциональность, плаксивость, раздражительность.

Диагностика
Врач может предположить заболевание по внешнему виду пациента. Однако высокий рост, худощавая комплекция и плохое зрение не всегда говорят о данной патологии. Эти признаки наблюдаются и при других генетических нарушениях, и у здоровых людей. Специалист обязательно расспрашивает больного о случаях этой наследственной аномалии в семье, в частности, у родителей.
Для диагностики синдрома Марфана необходимо пройти обследование у ортопеда, кардиолога, офтальмолога и генетика, а также сделать ЭКГ. Это поможет оценить состояние костей, глазного аппарата и сердца. Диагноз ставят, если у человека отмечается хотя бы один из следующих симптомов:
- деформация грудных костей;
- смещение хрусталика;
- искривление позвоночника;
- патологические изменения в аорте;
- удлинение и патологическая гибкость пальцев.
А также у человека при этом должны наблюдаться хотя бы два из следующих признаков:
- нарушение работы сердечных клапанов;
- пневмоторакс;
- плоскостопие;
- высокий рост;
- чрезмерная подвижность сухожилий и суставов;
- близорукость;
- растяжки на теле.
Таким образом, диагноз ставят по совокупности проявлений заболевания. Кроме этого, существует специальный анализ на синдром Марфана. У пациента берут кровь и проводят поиск мутаций в гене FBN1. Однако это довольно дорогостоящее исследование, его проводят в центрах молекулярной генетики.

Консервативное лечение
В современной медицине нет таких методов, которые могли бы исправить «поломку» гена. Поэтому лечение синдрома Марфана может быть только симптоматическим. Назначают следующие медикаменты, которые помогают скорректировать патологические изменения в организме:
- Бета-адреноблокаторы («Атенолол», «Обзидан» и другие). Их применяют при расширении аорты до 4 см для предотвращения кардиологических осложнений. Также используют и другие сердечные препараты (ингибиторы АПФ, антагонисты кальция).
- Препарат «Лозартан». Это лекарство стало использоваться в терапии болезни Марфана относительно недавно. Оно относится к блокаторам рецепторов ангиотензина. Препарат замедляет расширение аорты. По исследованиям медиков это лекарство действует более эффективно, чем бета-адреноблокаторы.
- Антибиотики и коагулянты. Их назначают обычно после кардиологических операций, для профилактики тромбов и эндокардита.
- Препараты для нормализации коллагена. С этой целью назначают антиоксиданты, витаминные комплексы и биодобавки: «Коллаген ультра», «Элькар», «Лимонтар», «Рибоксин», средства с коэнзимом, витамины Е и группы В.
- Ноотропы. Их назначают при слабости и астении. Чаще всего применяют «Пирацетам».
Больной должен находиться под наблюдением нескольких врачей (генетика, кардиолога, ортопеда, офтальмолога), регулярно проходить осмотры и необходимые обследования. Коррекция зрения при миопии проводится с помощью подбора очков или контактных линз.
Пациентам с болезнью Марфана показаны физиотерапевтические процедуры. Полезно проведение магнитотерапии на суставы, электросна, упражнений ЛФК. Раз в год рекомендуется лечение в санатории для больных с патологиями опорно-двигательного аппарата.
Хирургические операции
При этом синдроме часто приходится прибегать к кардиохирургическим вмешательствам. Это позволяет существенно увеличить выживаемость больных. До применения операций на сердце продолжительность жизни пациентов с этим синдромом была небольшой. Люди доживали только до среднего возраста (40-50 лет). В настоящее время кардиохирургическое лечение увеличило жизнь пациентов примерно на 20 лет.
Нередко приходится делать операции на аорте. Они показаны при расширении этого сосуда более 5 см. Также осуществляют протезирование митрального клапана.
Операции на сердце делают чаще всего при таком синдроме, так как этот орган сильно страдает от дефектов соединительной ткани. Однако проводят и другие хирургические вмешательства, такие как:
- Торакопластика. Эту операцию делают при деформации грудной клетки. Она позволяет избавиться от дефекта строения скелета. Проводится частичная резекция нескольких ребер, после этого грудина выглядит более ровной.
- Экстракция хрусталика. Такую операцию на глазах проводят при ранней катаракте и глаукоме.
- Стабилизирование позвоночника с помощью металлических пластин. Операцию проводят в тяжелых случаях сколиоза.
- Эндопротезирование. У больных часто отмечаются патологии костей. По этой причине иногда приходится заменять пораженный сустав на протез.
- Удаление аденоидов и гланд. Эти вмешательства необходимо провести в детстве. Ребенок, страдающий данным синдромом, очень подвержен простудным заболеваниям.
Кроме этого, беременным женщинам с болезнью Марфана показана операция кесарева сечения. Из-за слабости мышц и проблем с сердцем им очень трудно рожать самостоятельно.
Прогноз
При этом наследственном недуге необходимо комплексное лечение всех органов, которые затронуты болезнью. Без терапии продолжительность жизни больных составляет не более 40-45 лет. Смерть наступает от тяжелых нарушений работы сердца и сосудов.
Хирургическая коррекция патологических изменений позволяет значительно увеличить жизнь больных. При регулярном лечении и врачебном наблюдении пациенты могут дожить до пожилых лет. Кроме этого, после кардиохирургических операций возрастает трудоспособность больного.
Молодые женщины, страдающие этим синдромом, часто интересуются: можно ли им беременеть. Шанс родить здорового малыша существует при условии, что отец ребенка здоров. Однако необходимо помнить, что риск передачи заболевания в этом случае составляет 50%. К тому же вынашивание плода создает огромную нагрузку на сердечно-сосудистую систему, которая и так страдает при этом недуге. Поэтому при подобной генетической патологии беременеть не рекомендуется.
Профилактика
На сегодняшний день не разработана специфическая профилактика синдрома Марфана. Заболевание является генетическим и предотвратить его нельзя. Можно лишь пройти пренатальную диагностику во время беременности. Если один из будущих родителей страдает этой болезнью, то необходима консультация генетика.
Если у человека уже отмечаются признаки патологии, то необходимо избегать физических нагрузок. Больным не следует заниматься спортом и выполнять тяжелую работу. Такие пациенты должны регулярно проходить медицинское обследование. Дети с этим недугом освобождаются от занятий физкультурой, однако им показано выполнение упражнений ЛФК.
Источник