Фото детей с синдромом аперта

Синдром Апера представляет собой редкую генетическую патологию, выражающуюся в преждевременном внутриутробном закрытии костей черепа и дефектах костной системы. Клинически аномалия представлена необычной формой головы и лица, а также сращением пальцев на верхних (иногда и на нижних) конечностях. Патология встречается примерно у одного из 200 тысяч новорожденных.
Причины возникновения синдрома Апера
Симптомокомплекс, характерный для синдрома, впервые описал французский педиатр Эжен Апер (Аперт) в начале XX века. Аномалия также носит названия акрокраниодисфалангия, акроцефалосиндактилия, акросфеносиндактилия – все эти слова в переводе с латыни характеризуют отклонения от нормы в строении черепа, фаланг пальцев, описывая внешние признаки патологии.
Череп плода, находящегося в утробе матери, состоит из частей, которые ещё не полностью срослись между собой. Между теменными и лобными костями находится коронарный (венечный) шов, а между теменными – стреловидный (сагиттальный). Заращение этих соединительнотканных участков при описываемой аномалии происходит ранее, чем положено. Это приводит к появлению внешних признаков болезни и развитию внутренних патологий.

Мальчики с синдромом Апера рождаются также часто, как и девочки, то есть заболевание одинаково распространено среди представителей обоих полов. Основными факторами развития патологии считаются:
- Генная мутация. Изменение наследственных свойств в клетках организма одного из родителей в данном случае носит спонтанный характер. Ген, находящийся на десятой хромосоме и называемый FGFR2, расположен таким образом, что легко подвергается повреждениям. Именно этот участок ДНК отвечает за формирование и развитие соединительной (в частности, костной) ткани в эмбриональном периоде. Дефект гена приводит к заболеваниям костной системы. Вероятность рождения ребёнка с описываемым симптомокомплексом у пожилых родителей выше, чем у молодых.
- Наследственность. У человека, страдающего синдромом, с вероятностью 50% родится ребёнок с такой же аномалией.
- Рентгеновское облучение беременной женщины.
- Заболевания, перенесённые беременной: вирусы гриппа, туберкулёз, сифилис, краснуха и некоторые другие.
Синдром Апера, причины которого довольно сложно установить при отсутствии генетической предрасположенности, практически не поддаётся лечению.
Характерные симптомы патологии
Внешние признаки синдрома представляют чёткую клиническую картину, а в совокупности с другими проявлениями дают основание для постановки диагноза. Основные симптомы, определяемые у младенца в период новорожденности, вне зависимости от причин заболевания:
- Краниосиностоз – раннее сращение частей черепа – приводит к деформации черепных костей и определяет отклонение от нормы формы головы ребёнка. Такие дети имеют высокий выпуклый лоб, широкую переносицу, широко посаженные глаза, плоское лицо, выступающую нижнюю челюсть.
- Сращение пальцев на верхних и/или нижних конечностях перепончатое, костное либо кожное. Чаще всего соединёнными в процессе внутриутробного развития остаются указательный, средний и безымянный пальцы руки и аналогичные на ногах. Иногда три пальца срастаются настолько плотно, что на них формируется один общий ноготь.
- Дефекты внутриутробного развития костной ткани в ряде случаев приводят к карликовости.
- Позднее появление зубов, их деформация.
- Хроническая внутричерепная гипертензия, вызванная деформацией костей черепа, приводит к развитию слабоумия.
- У пациентов часто развиваются патологии внутренних органов: сердца, почек, поджелудочной железы. Нередко фиксируются аномалии развития позвонков, внешних половых органов. Страдает слух.
- Патологии органов зрения: косоглазие, вывих хрусталика, катаракта, нистагм, экзофтальм, пигментный ретинит и другие.
Совокупность клинических проявлений болезни позволяет сделать вывод о развитии синдрома Апера. На фото новорожденных, страдающих от патологии, видно сходство внешних черт.

Многие из описанных выше признаков разнятся в зависимости от условий жизни маленьких пациентов. Так, дети, от которых отказались родители, в детских учреждениях (детских домах, интернатах) показывают слабый уровень умственного развития. При этом ребёнок с диагнозом синдром Апера в возрасте 5 лет, живущий в семье, по уровню интеллекта может не уступать здоровым сверстникам.
Диагностика заболевания
Для постановки диагноза врачи изучают клиническую картину и данные анамнеза родителей. Из инструментальных методов применяют:
- Рентгенологические исследования. В младенческом возрасте показывают раннее закрытие швов черепа, во взрослом – деформацию черепных костей и позвоночника.
- Компьютерная томография черепа. Выявляет аномальное сращение костей (венечного шва), гипоплазию верхней челюсти, ненормальный размер глазниц и другие характерные признаки патологии.
- Магнитно-резонансная томография. Показывает степень изменения мягких тканей черепа.
- Ультразвуковые исследования плода. Некоторые отклонения внутриутробного развития (аномалии развития конечностей, костей черепа, пороки сердца) могут свидетельствовать о развитии синдрома Апера.
- Эхокардиография взрослых пациентов (УЗИ сердца) нередко показывает нарушения в работе сердечно-сосудистой системы: сердечную недостаточность, сужение просвета аорты и лёгочного ствола, дефект между правым и левым желудочками сердца.
- Молекулярно-генетический анализ ДНК. Высокоинформативный метод, позволяющий определить мутацию соответствующего гена.
Комплекс исследований позволяет провести дифференциальную диагностику с другими патологиями развития костной системы.
Корректная постановка диагноза имеет большое значение в связи с тем, что синдром Апера подразумевает лечение, отличное от терапии похожих заболеваний.
Лечение
Эффективного метода, который бы воздействовал на патогенез заболевания, не существует. Лечение носит паллиативный характер и направлено на минимизацию страданий пациента и улучшение качества жизни, насколько это возможно. Такую стратегию реализовывают посредством хирургического вмешательства и частично при помощи симптоматической медикаментозной терапии.

Операции при диагностированном синдроме выполняются по таким основным направлениям:
- Трепанация черепа. В процессе такой операции хирург производит разделение неправильно сросшихся частей черепа, восстанавливая его физиологическую форму. Хирургическое вмешательство иначе называется краниотомия, выполняется детям в возрасте около полугода. Оперативные манипуляции позволяют не только скорректировать форму головы, но и восстановить нормальное внутричерепное давление, а также избежать патологий, вызываемых гипертензией.
- Коррекция формы лица и глазного гипертелоризма. Операции преследуют преимущественно эстетические цели, проводятся в более позднем (от четырёх до двенадцати лет) возрасте.
- Хирургическое разделение пальцев. Производится в первые два года жизни ребёнка.
- Оперативное лечение сопутствующих симптомов (болезней сердца, органов зрения, слуха и так далее).
- Ортодонтические манипуляции с целью выравнивания прикуса и коррекции расположения и формы зубов.
Основную проблему – неправильно сросшиеся кости черепа, что провоцирует развитие черепной гипертензии – в современной медицине решают путём краниофациальной дистракции. Этот метод представляет собой постепенное растяжение костей черепа.
Медикаментозное лечение носит симптоматический характер. Пациентам назначают:
- Глазные капли и мази, снимающие патологическую сухость слизистых оболочек, а также лечащие сопутствующие патологии органов зрения.
- Препараты, снижающие внутричерепное давление.
- Антибиотики – против бактериальных инфекций верхних дыхательных путей, ушей, глаз, которым подвержены дети с синдромом Апера.
- Лекарственные средства, направленные на устранение сопутствующих симптомов, связанных с другими органами.
Эффективное лечение возможно лишь при условии слаженной работы врачей различных специализаций: хирурга, невролога, педиатра, офтальмолога, отоларинголога, психолога и других. Психическое здоровье пациента во многом определяется его адаптацией в социуме. Для решения этой проблемы привлекаются психотерапевты.
Возможные осложнения и прогноз
Вероятность развития осложнений на фоне отсутствия хирургического лечения высока. К наиболее вероятным из них относятся:
- развитие слабоумия;
- снижение зрения и слуха;
- заболевания позвоночника и суставов;
- патологии сердечно-сосудистой, выделительной и других систем.
Прогноз при отсутствии операции неблагоприятный, за исключением тех пациентов, которые дожили до средних лет, не имея пороков развития сердечно-сосудистой системы.
В случае своевременно проведённых оперативных вмешательств и лёгкой степени развития патологии пациент может жить до старости при условии, что мутация гена не повлекла за собой пороки сердца и тяжёлые патологии внутренних органов.
Профилактические рекомендации
Поскольку основной фактор развития синдрома – генные мутации, эффективной профилактики не существует. В данном контексте можно говорить о своевременной диагностике – УЗИ беременных женщин, особенно если в роду были случаи патологии.
Учитывая менее вероятные, но также существующие причины, к мерам профилактики можно отнести:
- недопущение рентгеновского облучения при беременности;
- своевременное лечение болезней при вынашивании плода.
Синдром Апера – редкая тяжёлая врождённая аномалия, не поддающаяся патогенетической терапии. Своевременное хирургическое и медикаментозное лечение носит паллиативный характер и может существенно улучшить качество жизни пациента.
Загрузка…
Источник
Что такое синдром Аперта?
Синдром Аперта (синдром Апера или акроцефалосиндактилия 1 типа) — редкое генетическое заболевание, возникающее при рождении. У больных с синдромом Аперта появляются характерные пороки развития черепа, лица, рук и ног.
Синдром Апера характеризуется краниосиностозом, состоянием, при котором фиброзные суставы (швы) между костями черепа преждевременно закрываются. Это может вызвать поражение лицевых костей и привести к тому, что верхняя часть головы будет выглядеть заостренной. При болезни может наблюдаться сращивание пальцев рук или ног. Пострадавшие дети также могут иметь интеллектуальные нарушения. Серьезность симптомов варьируется.
Синдром Аперта почти всегда является результатом новых генетических изменений (мутаций), которые происходят случайно. Редко болезнь наследуется по аутосомно-доминантному типу. Больные с синдромом могут проходить терапию, направленную на конкретные симптомы, включая реконструктивную хирургию черепа, лица, рук, ног.
Признаки и симптомы
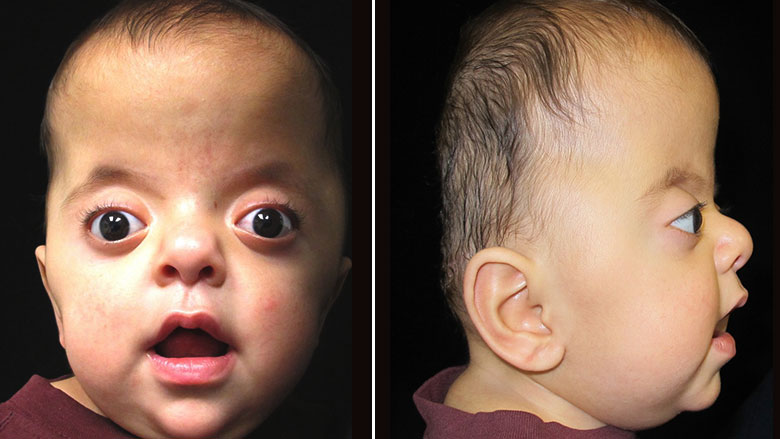
Синдром Аперта характеризуется краниосиностозом, преждевременным закрытием фиброзных суставов (швов) между определенными костями черепа. У детей без краниосиностоза швы позволяют голове расти и расширяться. Затем, эти кости сливаются воедино, образуя череп.
У людей с краниосиностозом мозг все еще растет после преждевременного закрытия этих швов.
Возникающее давление при росте головного мозга может привести к тому, что различные кости черепа и лица деформируются во время развития.
В зависимости от того, какие швы преждевременно закрываются, варьируется степень их тяжести. У большинства больных людей происходит преждевременное закрытие швов между костями, образующими лоб и верхнюю часть черепа. Это приводит к тому, что голова с рождения становится заостренной на верхней части (акроцефалия). Вдобавок, задняя часть черепа может выглядеть сплющенной, с высоким и широким лбом (см. фото выше). На черепе может быть большой «родничок» с поздним закрытием.
У некоторых больных также может быть гидроцефалия, при которой в полостях головного мозга накапливается спинномозговая жидкость. Это может вызвать внутричерепное давление.
Лицевые кости тоже могут быть поражены краниосиностозом. Это может привести к характерным дефектам лица. У пациентов с синдромом Аперта часто наблюдаются широко расставленные глаза (гипертелоризм), выпученные глаза (экзофтальм) или наклоненные вниз глазные щели. У них также могут быть слаборазвитые срединно-лицевые области (гипоплазия верхней челюсти) и расщелина нёба. Правая и левая стороны лица могут быть не симметричными.
Люди с болезнью могут иметь сплющенный нос с низкой переносицей. У людей бывает задержка роста зубов, скученность зубов или открытый прикус.
Если отверстия между носом и горлом сужены или заблокированы, или трахеальный хрящ поврежден, это может помешать дыханию и глотанию. Люди с этими проблемами могут иметь инфекции верхних дыхательных путей, апноэ во сне, недоедание.
Синдром Аперта имеет несколько характерных пороков развития кисти и стопы. У пострадавших людей могут быть короткие, широкие и большие пальцы ног, отклоняющиеся наружу. Они также могут иметь частичное или полное слияние (синдактилию) определенных пальцев рук и ног. Многие больные люди имеют полное слияние костей второго и четвертого пальцев и один единственный сплошной ноготь («варежкообразная» синдактилия). Однако возможны и другие слияния.
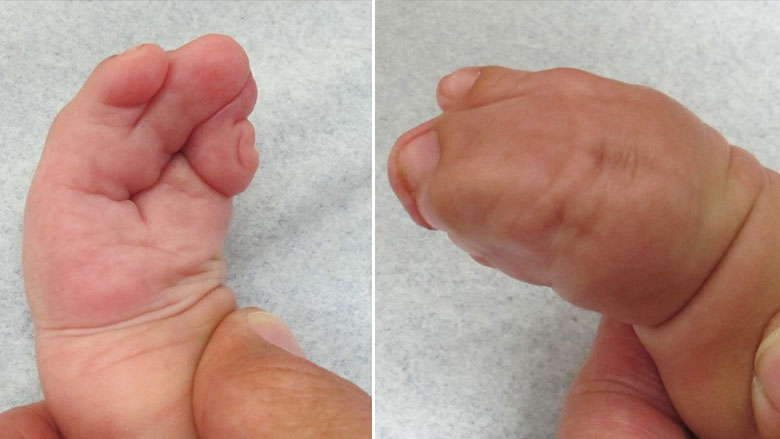
Суставы пальцев становятся жесткими к четырем годам. В ногах синдактилия также обычно затрагивает вторые, третьи и четвертые пальцы. Ногти на ногах могут быть частично непрерывными или отдельными. Как правило, синдром поражает верхние конечности сильнее, чем нижние.
Синдром Аперта может влиять и на другие системы органов (см. таблицу).
| Скелетная система |
|
| Нервная система |
|
| Уши |
|
| Сердечно-сосудистая система |
|
| Брюшная полость |
|
| Почки и мочеполовая система |
|
Причины
Синдром Аперта возникает в результате изменения (мутации) гена рецептора фактора роста фибробластов 2 (англ. Fibroblast growth factor receptor 2, сокр. FGFR2). Этот ген играет важную роль в развитии скелета. Гены предоставляют инструкции для создания белков, играющих разные роли в организме. Когда происходит мутация гена, белковый продукт может не работать должным образом. При синдроме Апера мутации в FGFR2 приводят к тому, что эти рецепторы неправильно связываются с факторами роста фибробластов. Это влияет на формирование нормальных швов в мозге и может препятствовать развитию многих других структур в организме. Это неправильное образование является причиной пороков развития, наблюдаемых при синдроме Аперта.
Почти у всех зарегистрированных пациентов расстройство было вызвано одной из двух специфических мутаций гена FGFR2. (Эти мутации обозначены как «Ser252Trp» и «Pro253Arg».) Эти мутации могут вызывать слегка отличающиеся проявления, включая степень выраженности синдактилии (сращивание пальцев). Различные мутации в гене FGFR2 могут вызывать несколько других связанных расстройств, включая синдром Пфайффера, синдром Крузона и синдром Джексона-Вейсса.
У 95% пациентов синдром Апера возникает в результате новой мутации в гене FGFR2. Эти новые мутации появляются случайно по неизвестным причинам (спорадически). Сообщалось, что отдельные случаи заболевания связаны с увеличением возраста отца.
Редко синдром наследуется по аутосомно-доминантному типу. Доминантные генетические нарушения возникают, когда необходима только одна копия мутации, чтобы вызвать конкретное заболевание. Риск передачи мутации от больного родителя к потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Синдром Аперта, по оценкам, встречается примерно у одного из 65 000 новорожденных. Мужчины и женщины болеют в относительно равных количествах. С тех пор, как расстройство было первоначально описано в 1894 и 1906 годах было зарегистрировано более 300 новых случаев. Сообщалось, что у людей из Азии самый высокий уровень заболеваемости данным синдромом.
Диагностика
Диагноз чаще всего ставится при рождении или в младенчестве. Пациенты диагностируются посредством клинической оценки и различных специализированных тестов. Определяются физические признаки, такие как аномалии лица или синдактилия.
Скелетные аномалии и врожденные пороки сердца обнаруживаются с помощью визуализации, например компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ). Нарушение слуха может быть обнаружено во время исследования слуха у новорожденного.
Люди могут также пройти тест на мутации в гене FGFR2, что может обеспечить генетическим диагнозом синдрома Апера.
В некоторых случаях признаки синдрома Аперта могут быть обнаружены до рождения. Это можно сделать с помощью пренатального 2D или 3D ультразвукового исследования или магнитно-резонансной томографии (МРТ). Ультразвуковое исследование (УЗИ) — неинвазивная процедура, позволяющая получить изображение плода. Процедура может обнаружить различия в форме черепа, аномалии лица и синдактилию. МРТ плода может обеспечить большую детализацию мозга плода, чем УЗИ.
Схожие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Аперта. Сравнения могут быть полезны для дифференциальной диагностики.
- Синдром Карпентера — редкое генетическое заболевание, связанное с краниосиностозом, полидактилией или синдактилией. Макушка головы может казаться необычно заостренной (акроцефалия) или голова может казаться короткой и широкой (брахицефалия). Вдобавок, черепные швы часто сливаются неравномерно, в результате чего голова и лицо кажутся асимметричными от одной стороны к другой. В некоторых случаях присутствуют дополнительные физические отклонения, такие как низкий рост, врожденные пороки сердца, легкое или умеренное ожирение, пупочная грыжа или крипторхизм. Многие люди с расстройством страдают от легкой до умеренной умственной отсталости.
- Синдром Крузона — редкое наследственное заболевание, связанное с краниосиностозом. У людей с синдромом Крузона также имеются пороки развития средней части лица, выпуклые глаза и закупорка дыхательных путей, приводящие к затруднению дыхания и глотанию. У некоторых больных голова очень большая (гидроцефалия). Синдром Крузона обычно не связан с умственной отсталостью или проблемами затрагивающими руки, ноги, кисти или ступни. Синдром Крузона вызывается изменениями в одном из генов FGFR, обычно FGFR2, и наследуется по аутосомно-доминантному типу.
- Синдром Джексона-Вейсса (СДВ) — редкое наследственное заболевание, характеризующееся краниосиностозом и аномалиями ног. Диапазон и серьезность симптомов и признаков сильно различаются, даже среди затронутых членов одной семьи. Первичные признаки могут включать необычно плоские, недоразвитые срединно-лицевые области (гипоплазия средней части лица), аномально широкие большие пальцы ног и/или порок развития или слияние определенных костей в ступнях. СДВ может возникать спорадически или наследоваться по аутосомно-доминантному типу.
- Синдром Пфайффера — редкое генетическое заболевание, характеризующееся краниосиностозом и аномально широкими и отклоненными медиально большими пальцами рук и ног. У большинства пострадавших также наблюдаются выпученные глаза и кондуктивная потеря слуха. Существует три формы синдрома Пфайффера. Типы II и III являются более серьезными. Синдром Пфейффера является аутосомно-доминантным расстройством, связанным с мутациями в генах FGFR2 и FGFR1.
Лечение
Лечение синдрома Аперта варьируется в зависимости от того, какие симптомы наблюдаются у больного. Лечение может потребовать ухода со стороны группы медицинских специалистов, включая педиатров и хирургов, нейрохирургов, врачей, специализирующихся на заболеваниях скелета, суставов и мышц (ортопедов), врачей, специализирующихся на заболеваниях ушей, носа и горла (отоларингологов), врачей, специализирующихся на нарушениях сердечной деятельности (кардиологов).
Специальные методы лечения синдрома Аперта являются симптоматическими и поддерживающими. Краниосиностоз и гидроцефалия могут привести к ненормально повышенному давлению внутри черепа и мозга. В таких случаях для коррекции краниосиностоза может быть рекомендовано раннее хирургическое вмешательство (в течение 2-4 месяцев после рождения). Для пациентов с гидроцефалией операция может также включать введение трубки (шунта) для оттока избыточной спинномозговой жидкости (СМЖ) из головного мозга. СМЖ сливается в другую часть организма, где он абсорбируется.
Корректирующая и реконструктивная хирургия может быть рекомендована для коррекции черепно-лицевых пороков развития. Хирургия также может помочь исправить полидактилию и синдактилию, а также другие скелетные дефекты или физические отклонения. Для больных с врожденными пороками сердца может потребоваться лечение определенными препаратами, хирургическое вмешательство и/или другие терапевтические меры. Для некоторых больных с нарушениями слуха полезны слуховые аппараты.
Раннее вмешательство может быть важным для обеспечения того, чтобы дети с синдромом Аперта полностью раскрыли свой потенциал. Специальные терапевтические мероприятия, такие как физиотерапия, трудотерапия и специализированное обучение также будут полезными.
Для пострадавших лиц и их семей рекомендуется генетическое консультирование. Генетический консультант сможет объяснить причины заболевания. Он также может обсудить возможность рождения новых детей с заболеванием. Дополнительно, для всей семьи необходима психосоциальная поддержка.
Прогноз
Прогноз для детей с синдромом Апера зависит от того, насколько серьезным является состояние и на какие системы организма оно повлияло. Заболевание может быть более серьезным, если повлияет на дыхательную систему ребенка или если внутри черепа вырастит давление, но эти проблемы можно исправить хирургическим путем.
Дети с синдромом часто имеют проблемы с обучением. Некоторые дети страдают более серьезно, чем другие.
Поскольку тяжесть болезни может варьироваться в широких пределах, сложно прогнозировать продолжительность жизни. Заболевание может не оказывать существенного влияния на продолжительность жизни ребенка, особенно если у него нет пороков сердца.
Источник