Фокальная дермальная гипоплазия синдром гольтца

а) Эпидемиология. Фокальная дермальная гипоплазия Гольтца (ФДГ), впервые описанная в 1934 г., получила свое название благодаря описанию Гольтцем и соавт. троих пациентов. В дальнейшем было описано более 200 зарегистрированных случаев болезни.
б) Этиология, патогенез, генетика. Фокальная дермальная гипоплазия представляет собой Х-сцепленное доминантное заболевание, которое обычно имеет летальное течение у пациентов мужского пола. Полагают, что клинические случаи с описанием больных мужского пола связаны с мозаицизмом соматических мутаций (этот факт подтвержден для синдрома недержания пигмента); выживание больных мальчиков обеспечивает наличие нормальных клеток.
Ген, в котором развивается мутация — PORCN, представляет собой человеческий гомолог гена porcupine у дрозофил. Считается, что PORCN важен для процесса пальмитоиляции и секреции белка Wnt, основного регулятора развития кожи и костей.
в) Клиника:
1. Кожные проявления. Основными диагностическими признаками при фокальной дермальной гипоплазии Гольтца (ФДГ) являются кожные изменения. К ним относятся линейная, точечная, полосатая решетчатая атрофии с телеангиэктазиями. Решетчатая атрофия представляет собой мелкие точечные вдавления в коже, которые располагаются вдоль линий Блашко.
Участки истонченной или отсутствующей кожи неравномерно распределены по поверхности тела, вследствие чего выпячивания жировой ткани имеют вид желто-розовых разрастаний над поверхностью кожи. Эти разрастания легко сдавливаются. Папилломы, которые могут быть мягкими или сосудистыми, развиваются на протяжении всей жизни и чаще поражают перигениталь-ные, периоральные, интертригинозные участки и слизистые оболочки.
К другим кожным симптомам относятся очаговая алопеция, ломкие или редкие волосы, а также гиперкератоз ладоней и ступней. У некоторых пациентов отмечался гипергидроз, еще у части пациентов была выявлена врожденная аплазия кожи.
2. Сочетанные системные проявления. В патологический процесс при фокальной дермальной гипоплазии Гольтца (ФДГ) также вовлекаются другие органы и системы, наиболее часто это костная, центральная нервная система, зубы и глаза. Типичным является поражение глаз в виде микрофтальма и колобомы, поэтому больным с диагнозом ФДГ необходимо проводить полное офтальмологическое обследование.
Также распространены олигодонтия, дисплазия зубов и дефекты зубной эмали. Перечень костных аномалий слишком велик, чтобы перечислять все эти симптомы; наиболее часто встречаются полосчатая остеопатия (образование вертикальных полосок на костях), синдактилия (как кожная, так и костная), асимметрия и низкорослость. Примерно в 15% случаев выявляется задержка умственного развития. В небольшом количестве случаев были описаны дефекты других органов и систем, в том числе — аномалии сердца, передней брюшной стенки и почек. Совершенно необязательно, что эти симптомы являются неотъемлемыми компонентами синдрома фокальной дермальной гипоплазии Гольтца (ФДГ).
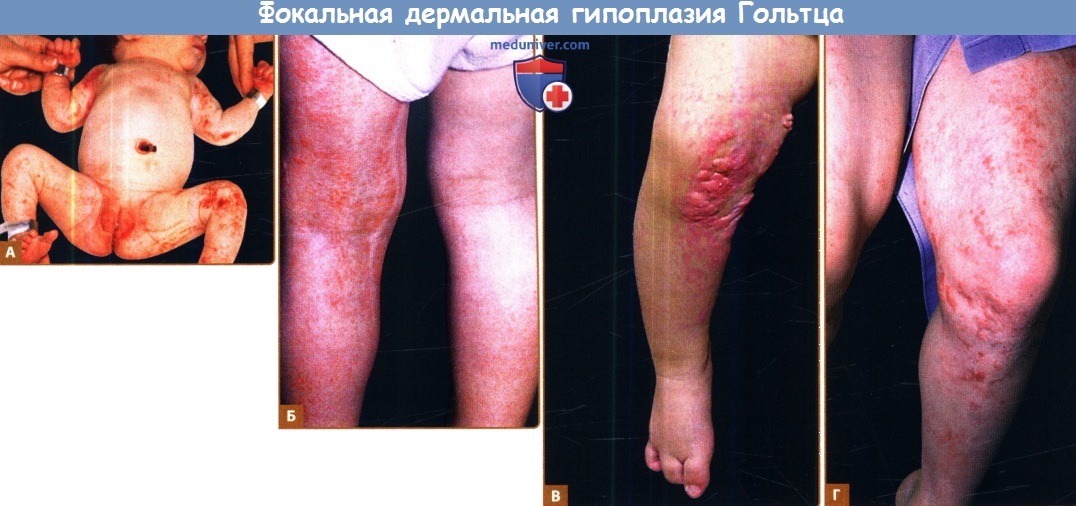
Фокальная дермальная гипоплазия Гольтца.
А. Полосчатое и очаговое поражение кожи новорожденного. Обратите внимание на сходность в распространении патологических элементов с синдромом недержания пигмента.
Б, В. Та же девочка в возрасте двух лет с атрофией и выпячиваниями жировой ткани.
Г. Молодой человек, у которого преобладают эритема и атрофия с сухостью и образованием чешуек.
г) Диагностика и дифференциация. Диагностика фокальной дермальной гипоплазии Гольтца (ФДГ) относительно проста. Решетчатая атрофия была описана также при Х-сцепленном доминантном синдроме Коранди-Хюнерманна (точечная хондродисплазия), в отличие от которого при фокальной дермальной гипоплазии Гольтца не встречается ихтиоз, и при котором, в отличие от фокальной дермальной гипоплазии Гольтца, не бывает выпячиваний подкожно-жировой клетчатки.
Полосчатое распространение атрофических участков, точно так же, как и другие системные проявления, могут напоминать синдром недержания пигмента, но характерные для недержания пигмента образование пузырей, гиперкератоз и гиперпигментация, при фокальной дермальной гипоплазии Гольтца не выявляются. При микрофтальме и линейных кожных дефектах с микрофтальмом, дермальной аплазией и склерокорнеа кожные проявления ограничены областью головы и шеи; отмечается атрофия и рубцевание кожи, более сходное с врожденной аплазией кожи, но не с дермальной атрофией. Заболевания имеют сходные глазные проявления.
д) Лечение и прогноз. Специфических методов лечения кожных и системных проявлений фокальной дермальной гипоплазии Гольтца нет. Папилломы могут удаляться, если они нарушают функции органа. Хороший косметический эффект может давать применение сосудистых лазеров для уменьшения эритемы в телеангиэктатических областях. Как и при большинстве Х-сцепленных доминантных заболеваний, степень клинических проявлений и тяжести течения болезни значительно варьирует.
Это затрудняет прогнозированрре заболевания в раннем грудном возрасте, поэтому обычно разумнее оставлять составление прогнозов на будущее, когда системные проявления станут более очевидными. Важны сведения о прерванных беременностях (измененное соотношение детей мужского пола к детям женского пола и повышенная частота прерванных беременностей позволяют предположить, что мать является носителем мутации). Оба родителя должны проходить тщательное обследование; у отцов могут отмечаться легкие симптомы болезни, предположительно они являются носителями постзиготных мутаций гена FDH.
— Рекомендуем далее ознакомиться со статьей «Признаки наследственных иммунодефицитов и их классификация»
Редактор: Искандер Милевски. Дата публикации: 25.12.2018
Источник
 Синдром, описанный Гольтцом в 1962 году, представляет многопорочное сочетание, последствие некоторых аномалий экто- и мезодермального развития, поражающих, в особенности, кожу, мышцы, скелет и глаза.
Синдром, описанный Гольтцом в 1962 году, представляет многопорочное сочетание, последствие некоторых аномалий экто- и мезодермального развития, поражающих, в особенности, кожу, мышцы, скелет и глаза.
Появляется у детей с рождения. До 1970 г. было опубликовано только 30 клинических наблюдений.
Этиопатогенез синдрома Горлина—Гольтца.
Этиология неизвестна. Генотипический характер синдрома вероятен, но не доказан. Кариотип—нормальный. Синдром появляется преимущественно у детей женского пола.
Синдром Горлина—Гольтца с клинической точки зрения похож на синдром Ротмунда, носящий выраженный семейный и наследственный характер, передающийся рецессивно-аутосомным образом и так же преимущественно появляющийся у женского пола.
Этот синдром известен в литературе под другими названиями, а именно:
- фокальная дермальная гипоплазия;
- синдром Гольтца-Петерсона-Горлина-Бавитца;
- синдром FDH;
Симптоматология синдрома Горлина—Гольтца
Кожные явления. У новорожденных кожные поражения могут проявляться в 3 клинических формах:
- Эритематозные, неправильной формы зоны, расположенные на туловище и на конечностях; позже эритематозные поражения становятся бледными и стертыми;
- Псевдобулезный аспект в некоторых небольших зонах кожи.
- Небольшие или более обширные зоны кожной врожденной аплазии, расположенные неравномерно, преимущественно на туловище и на конечностях.
Эти зоны могут чередоваться с некоторыми выпуклыми поражениями на поверхности кожи или даже с небольшими липоматозными опухолями. Со временем, зоны кожной аплазии превращаются в зоны с остаточными явлениями кожной атрофии или в зоны дермальной атрофии с липоматозом. В пораженных зонах появляется обычно гиперпигментация, однако, с одновременным наличием и зон гипопигментации.
У детей раннего возраста и дошкольника кожные поражения отличаются от новорожденного, проявляясь также в 3 клинических формах.
- Круглые или овальные зоны, с диаметром в несколько сантиметров, цвета охры или светло-желтого цвета из-за развивающегося липоматоза на здоровой кожной ткани, или в апластических кожных зонах, оставшихся после периода новорожденности. Чаще всего, они локализованы на туловище и конечностях.
- Участки дермальной гипоплазии, сочетающиеся с телеангиэктазией и с пигментными пятнами разных размеров (от нескольких мм до нескольких см), расположенными в виде зон, либо линейно в виде ленты.
- Ангиофиброматозный папилломатоз вокруг отверстий, локализованный, в особенности, на околоротовых и околоанусовых покровах или на слизистой миндалин и мягкого неба.
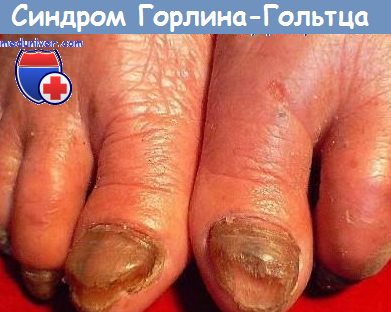
Кожные придатки сильно изменены. Отмечается: очаговая алопеция; ангидроз; искривление ногтей, хрупкость и в некоторых случаях даже их отсутствие.
Аномалии зубов: зубы небольших размеров, патологической формы или с порочной посадкой, иногда гипоплазия зубной эмали.
Аномалии скелета и мышц: синдактилии очень разнообразного типа и сочетания, проявляющиеся чаще всего только как кожные синдактилии; когда затронуты и кости, синдактилии появляются в особенности на III и IY пальцах обеих конечностей. В другом случае, синдактилии сочетаются с полидактилией или клинодактилией (в некоторых случаях последние представляют единственные аномалии пальцев). Гораздо реже отмечается аплазия или гиплоплазия разных пальцев (олигодактилия); сколиозы; genu valgum.
Глазные аномалии — разные (существуют приблизительно в 50% случаев), все ведут к тяжелым расстройствам зрения. Встречаются: микрофтальмия, часто в сочетании с микроцефалией; коллобома ириса и хороида; отсутствие ириса; подвывих хрусталика, косоглазие.
Патологическая анатомия синдрома Горлина—Гольтца.
Во всех случаях эпидермис не поражен. Характерные поражения локализованы в дерме. Структура дермы совершенно изменена, появляются гипопластические зоны, сопровождающиеся местными жировыми и ангиоматозными изменениями соединительной ткани: коллаген дермы пропитан интерстициальной жидкостью.
Течение и прогноз синдрома Горлина—Гольтца
Клиническое течение, благоприятное с точки зрения кожных проявлений; после выздоровления выявляются лишь малозначительные остаточные явления в дерме. Глазные признаки, без тенденции к регрессу, могут закончиться окончательным пороком.
Лечение синдрома Горлина—Гольтца.
При синдроме Гольтца не существует эффективного лечения, как и при всех других глазных и кожных синдромах, обусловленных аномалиями экто- и мезодермы. Генетический совет может оказаться полезным в жизни этих больных. Врач обязан информировать семью о наследственном характере аномалии и привлечь внимание родителей на возможность ее появления у наследников.
Источник
29.06.2010г.
Синоним: синдром Гольтца. Заболевание описано в 1962 г. R. Goltz с соавт.
Минимальные диагностические признаки: участки истонченной кожи.
Клиническая характеристика
Кожные изменения характеризуются обширными сетчатыми или линейными участками истонченной кожи с выпячиванием жировой клетчатки (100%), полным отсутствием кожного покрова в некоторых участках тела, пигментными или депигментированными полосами, телеангиэктазиями, папилломами на губах, деснах, на основании языка, во влагалище. Папилломы также могут отмечаться в паховой, подмышечной и около пупочной областях.
Поражение кожи может проходить несколько стадий:воспалительную, десквамативную, пузырчатую, уртикарную или стадию выраженной эритемы. Отмечаются фолликулярный гиперкератоз, папулезные изменения, редкие ломкие волосы, отсутствие или дистрофия ногтей.
Наблюдаемые дефекты скелета: низкий рост, микрокрания, округлая форма черепа, заостренный подбородок, истончение и искривление носовой перегородки, прогнатизм, кифоз, сколиоз, слияние и сакрализация позвонков, скрытая расщелина позвоночника, рудиментарный хвост, асимметрия лица, туловища и конечностей, пороки развития дистальных отделов конечностей (гипоплазия или отсутствие пальцев, полигактилия, клешневидная кисть, синдактилия, слияние фаланг, камптодактилия, клинодактилия, вальгусная деформация), генерализованный остеопороз.
Наблюдаются анофтальмия, микрофтальмия, гипертелоризм, аниридия, нистагм, косоглазие, гетерохромия и колобома радужки, голубые склеры, подвывих хрусталика, колобома сосудистой оболочки, сетчатки и зрительного нерва, де- или гиперпигментация радужки, помутнение роговицы и стекловидного тела, эктропион века, птоз, закупорка слезных каналов.
Отмечаются недоразвитие нижней челюсти, аномалии прикуса, микродонтия, дисплазия и агенезия зубов, аномальное расположение зубов, дефекты эмали и кариес, расщелина губы, высокое арковидное небо, дефект альвеолярных отростков, срединная расщелина языка, двойная уздечка языка, гемигипоплазия языка, гипертрофия десен.
Имеют место недоразвитие крыльев носа, выступающие асимметричные ушные раковины, гипоплазия завитка, преаурикулярные выросты, смешанная глухота, шейные фистулы, гипоплазия I и V пальцев кисти, расхождение прямых мышц живота, омфалоцеле, паховая и пупочная грыжа, выпадение прямой кишки, асимметрия молочных желез с латеральным расположением ареол, пороки сердца (дефекты межпредсердной перегородки, стеноз легочного ствола), аномалии почек и мочеточников. Характерна умственная отсталость. Популяционная частота неизвестна.
Соотношение полов — М0:Ж1.
Тип наследования — Х-сцепленный доминантный.
Дифференциальный диагноз: врожденная пойкилодермия, синдром недержания пигмента, синдром эпидермальных невусов.
«Наследственные синдромы и медико-генетическое консультирование»,
С.И. Козлов, Е.С. Еманова
Читайте далее:
- Пикнодизостоз (pycnodysostosis)
- Почек, гениталий и среднего уха аномалии (renal, genital and middle ear anomalies)
- Рассела-Сильвера синдром (Silver-Russell syndrome)
- Рубинштейна-Тейби синдром (Rubinstein-Taybi syndrome)
- Спондилоэпифизарная дисплазия поздняя (spondyloepiphyseal dysplasia tarda)
- Триптофана мальабсорбция (tryptophan malabsorption)
- Фетальный цитомегаловирусный синдром (fetal cutomegalovires syndrome)
- Хондродисплазия точечная, тип Конради-Хюнермана (chondrodysplasia punctata, Conradi-Hunermann type)
- Хромосомы llp+ синдром (chromosome llp+ syndrome)
- Хромосомы X моносомии синдром (turner syndrome)
- Эктодермальная дисплазия ангидротическая (ectodermal dysplasia anhidrotic)
- Эритрокератодермия вариабельная (erythrokeratodermia variabilis)
- Пилоростеноз (pyloric stenosis)
- Почечный канальцевый ацидоз (renal tubular acidosis)
- Расщелина губы с или без расщелины неба (cleft lip with or without cleft palate)
- Секкеля синдром (Seckel syndrome)
- Срединной расщелины лица синдром (median cleft face syndrome)
- Трихо-рино-фалангеальный синдром, тип I (tricho-rhino-phalangeal syndrome, type I)
Источник
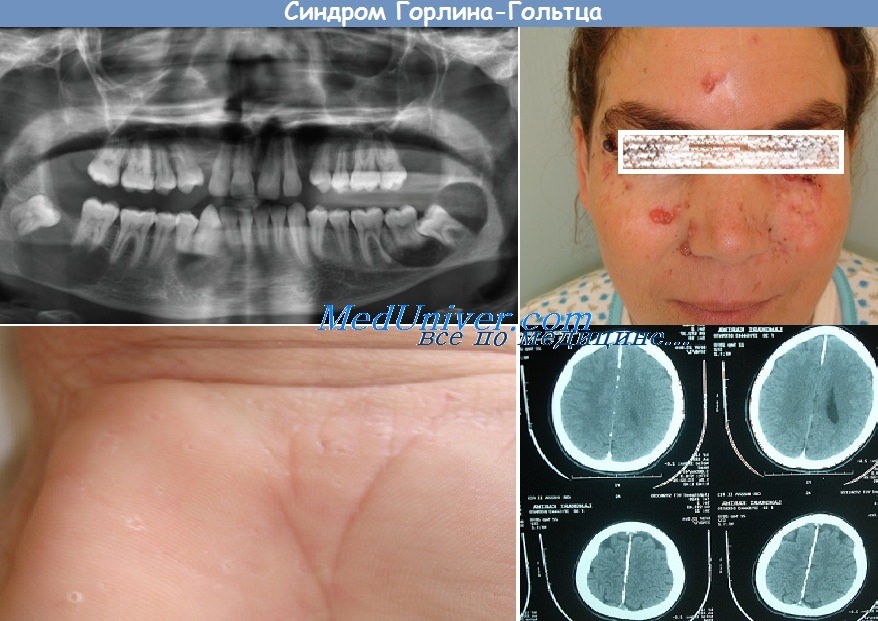
Синдром Горлина—Гольтца. Диагностика и лечение синдрома Горлина-Гольтца.Синдром Горлина—Гольтца (син.: синдром невоидной базальноклеточной карциномы, синдром базальноклеточных невусов) — генетически детерминированный полиорганный синдром, наследуемый по аутосомно-доминантному типу с высокой пенетрантностью и различной экспрессивностью. Мутантный ген локализуется в хромосоме 9 на участке q—22.3-q31. Основное проявление синдрома — множественные базалиомы, которые ассоциируются с разнообразными пороками развития скелета, глаз, нервной, эндокринной систем и других органов и тканей, а также с опухолями различной локализации. Заболевание обусловлено мутациями гена PTSH в хромосоме 9 и ассоциируется с антигенами Н1А-А10, В14. Почти все изменения врожденные. Базалиомы также могут быть врожденными, хотя чаще появляются в позднем детском возрасте, как правило они развиваются у лиц не старше 35 лет. Обычно это множественные базалиомы, располагающиеся симметрично и билатерально как на открытых, так и на закрытых участках кожи. Преимущественно поражаются лицо, шея, туловище и конечности. Количество базалиом может достигать нескольких сотен. Вначале процесс представлен поверхностными базалиомами диаметром от 1 до 3 см, развивающимися медленно и практически не меняющимися по величине до наступления 2-го или 3-го десятилетия жизни. Затем отдельные новообразования могут быстро увеличиваться до 5-10 см. изъязвляться, под влиянием неблагоприятных внешних факторов среди них появляются кистоз-ные, язвенные формы, а также метатипический рак кожи. Точечные пигментированные углубления на ладонях и подошвах нередко возникают раньше других кожных проявлений синдрома и встречаются в 70-80% случаев, на их дне почти всегда имеются телеангиэктазии. Количество элементов — до нескольких сотен, больше всего их на боковых поверхностях ладоней, подошв и пальцев кистей. Они возникают из-за преждевременного слущивания рогового слоя эпидермиса. Изредка из углублений в области ладоней и подошв могут развиваться опухоли. Синдром Горлина—Гольтца также может ассоциироваться с другими поражениями кожи: эпидермальными кистами, милиумом, фибромами, липомами, пигментными невусами, веснушками, ладонно-подошвенным гиперкератозом, комедонами . Лишь у отдельных больных с синдромом Горлина—Гольтца отсутствуют кожные опухоли. Среди различных костных аномалий, которые имеют врожденный характер и выявляются у 75-90% больных с этим синдромом, наиболее характерны множественные одонтогенные кисты верхней и нижней челюстей которые при нагноении приводят к отечности и тупой боли и могут вскрываться в полость рта. Описываются также кифоз, сколиоз, расщепление ребер, воронкообразная грудная клетка, укорочение IV пястных костей, синостозы, прогнатия, парадонтоз, неправильное расположение зубов, готическое нёбо, срединный носовой синус, широкие носовые ходы, истинный гипертелоризм, выступающие лобные бугры, дизостозы костей лицевого черепа, субкорнеальные кистозные изменения длинных трубчатых и плоских костей. Аномалии глаз встречаются у 26% больных и проявляются врожденной слепотой, гипертелоризмом, дистопией внутренних углов глаза, катарактой, глаукомой, колобомой, стробизмом.
Поражения центральной нервной системы отмечаются у 10-42% больных и проявляются гидроцефалией, микроцефалией, недоразвитием мозолистого тела головного мозга, пластинчатым обызвествлением серпа большого мозга, кальцификацией твердой и мягкой мозговых оболочек, эпилепсией, деменцией, задержкой психического развития. Патология со стороны эндокринной и половой систем (крипторхизм, гипогонадизм, бесплодие, двурогая матка, фиброматоз яичников, акромегалия, тиреоидит) наблюдается у 17% больных. Синдрому Горлина— Гольтца могут сопутствовать и другие аномалии развития: врожденное отсутствие почки и мочеточника, анемия Фанкони и др. Синдром Горлина— Гольтца ассоциируется с такими опухолями, как медуллобластома, фиброматоз яичников, фибросаркомы челюстей, фибромы, тератомы и цистаденомы яичников, лейомиомы, нейрофиброматоз I типа, болезнь Ходжкина, ретинобластома, менингиома, рабдомиома, множественные миомы матки, семинома, рак матки, ренинсекретирующая опухоль яичника, опухоль мозжечка, менингиома. В частности, медуллобластома (мутации в гене 9q.31) чаще поражает мальчиков в возрасте до 2 лет, в связи с чем нередко возникает до появления кожных опухолей. Другие опухоли при синдроме Горлина—Гольтца, в отличие от первично-множественных базалиом, протекают более агрессивно и резистентно по отношению к проводимой терапии, часто рецидивируют и даже могут метастазировать. Диагноз синдрома Горлина— Гольтца устанавливается на основании совокупности клинико-гистологических данных и результатов обследования стоматологом, челюстно-лицевым хирургом, невропатологом, окулистом, эндокринологом, гинекологом; обязательна рентгенография плоских костей, а для выявления пластинчатого обызвествления серпа большого мозга — рентгенологическое исследование черепа. Дифференциальный диагноз проводят с первично-множественными базалиомами. Течение синдрома Горлина— Гольтца сопровождается появлением новых опухолей на протяжении всей жизни. Прогноз лучше, чем при медуллобластоме, возникающей независимо от синдрома Горлина—Гольтца. Опухоли, расположенные в центральной части лица, на ушных раковинах или в их окружности, подлежат иссечению по методу Mohs — с микроскопией замороженных горизонтальных срезов для определения объема операции. Мелкие опухоли на туловище и конечностях удаляют путем электрокоагуляции или проводят комбинированное лечение этретинатом и флуороурацилом (5% крем) в течение 25-30 сут. В профилактических целях используют этретинат.
— Также рекомендуем «Нейрофиброматоз. Причина и признаки нейрофиброматоза.» Оглавление темы «Генетически обусловленные кожные заболевания.»: |
Источник